提取物和浸出物(Extractables and Leachables�����,簡稱E&;L)相容性研究是確保藥品包裝材料����、設(shè)備與藥品制劑在儲存和使用過程中不發(fā)生有害相互作用的重要工作。美國FDA高度重視E&L評估����,在藥品上市申請審查中頻繁提出關(guān)于E&L研究的補充信息請求(發(fā)補意見)����。為了幫助企業(yè)更好地理解和應(yīng)對這些常見問題,本文梳理了FDA對E&L研究的主要發(fā)補意見類型��,并逐一解讀其含義和應(yīng)對建議����。
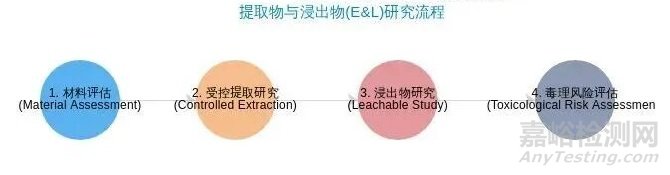
首先,我們可以通過以下圖表直觀地了解E&L研究的基本流程和FDA關(guān)注的關(guān)鍵環(huán)節(jié)��。
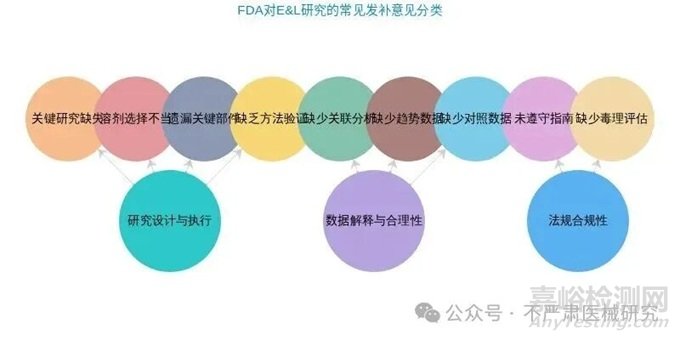
常見發(fā)補意見分類與示例
FDA在審評過程中針對E&L研究的發(fā)補意見主要集中在研究設(shè)計與執(zhí)行��、數(shù)據(jù)解釋與合理性����、法規(guī)合規(guī)性等幾個方面。下圖展示了這些主要的發(fā)補意見分類及其包含的具體內(nèi)容����。
下面將分別介紹各類常見問題����,并通過實例進(jìn)行解讀����。
1. 研究設(shè)計與執(zhí)行方面的不足
此類問題主要涉及E&L研究在實驗設(shè)計、執(zhí)行和覆蓋范圍上的缺陷�����。FDA要求研究設(shè)計全面����、合理,涵蓋所有關(guān)鍵材料和條件����,以確保評估的完整性。以下是常見的設(shè)計執(zhí)行類不足:
關(guān)鍵研究缺失或不足: 研究方案遺漏了重要的測試或分析��。例如��,許多補充申請缺乏對主要包裝系統(tǒng)浸出物的充分研究�����。FDA期望E&L研究應(yīng)詳細(xì)分析包裝材料中的雜質(zhì)、添加劑和降解產(chǎn)物��,評估其對藥品的潛在影響��。如果提交資料中未包含這些研究����,F(xiàn)DA可能要求補充完整的包裝相容性數(shù)據(jù)��。
溶劑選擇不當(dāng): 提取物研究中使用的溶劑范圍有限����,未能充分模擬實際情況。FDA強調(diào)應(yīng)采用不同極性的多種溶劑進(jìn)行嚴(yán)格的提取物研究����,以識別廣泛的潛在遷移物。如果溶劑選擇不全面����,可能導(dǎo)致某些可提取物未被檢測到,從而遺漏風(fēng)險�����。因此,企業(yè)需要確保在提取物研究中使用足以溶解各種極性成分的溶劑體系�����。
遺漏關(guān)鍵部件評估: 研究未包括所有與藥品直接接觸的關(guān)鍵部件�����。例如��,制造過程中使用的材料(如硅膠管�����、管道等)未進(jìn)行E&L評估��。這些組件可能在灌裝過程中引入雜質(zhì)����,影響藥品質(zhì)量。又如����,某些特殊組件(如包裝上的淋膜、標(biāo)簽黏合劑等)未被納入相容性評估。FDA指出�����,必須全面覆蓋所有與藥品接觸的材料����,包括制造設(shè)備和包裝附件,否則可能存在未被發(fā)現(xiàn)的安全隱患��。
缺乏驗證的分析方法: 研究中使用的分析方法未經(jīng)過充分驗證����。例如�����,方法的檢出限和定量限未滿足要求����,導(dǎo)致某些低水平的浸出物未被檢測到。又如��,方法的特異性不足��,無法區(qū)分背景干擾與真正的浸出物。FDA可能要求企業(yè)提供方法驗證報告�����,證明分析方法的可靠性����,確保所有可能的浸出物都能被準(zhǔn)確檢測和定量。
研究條件不充分: 實驗條件設(shè)置不全面��,未能覆蓋最壞情況或關(guān)鍵場景��。例如����,溫度和時間設(shè)置不夠極端,不足以揭示長期儲存或使用過程中的遷移風(fēng)險�����。再如�����,未考慮藥品在不同使用條件下的情況(如輸液過程中的熱暴露或長時間接觸)�����。FDA期望E&L研究應(yīng)模擬最嚴(yán)格的儲存和使用條件,包括加速老化條件��,以確保藥品在整個生命周期內(nèi)的安全性��。因此����,企業(yè)應(yīng)確保研究設(shè)計覆蓋溫度、時間�����、劑型等關(guān)鍵參數(shù)的最壞情況�����,并在必要時進(jìn)行額外的加速穩(wěn)定性研究�����。
示例解讀: 某仿制藥申請(ANDA)在提交資料中未提供任何關(guān)于玻璃容器和橡膠塞浸出物的研究數(shù)據(jù)����。FDA據(jù)此提出發(fā)補,要求申請人補充完整的E&L研究�����,包括在模擬藥品配方的條件下評估容器和密封件釋放的物質(zhì)��。申請人隨后補充了相關(guān)研究��,結(jié)果顯示浸出物水平符合安全閾值�����。該案例說明�����,研究設(shè)計的完整性對獲得FDA認(rèn)可至關(guān)重要����。
2. 數(shù)據(jù)解釋與合理性方面的不足
此類問題涉及對E&L研究數(shù)據(jù)的分析、解釋和支持理由的不足����。FDA要求申請人在研究報告中清晰地闡述數(shù)據(jù)含義,并提供科學(xué)依據(jù)證明其安全性�����。以下是常見的數(shù)據(jù)解釋類不足:
缺乏提取物與浸出物的關(guān)聯(lián)分析: 研究中提取出的化合物未與實際產(chǎn)品中的浸出物建立關(guān)聯(lián)。FDA希望看到提取物分析結(jié)果與浸出物測試結(jié)果之間的比較����,以了解長期儲存過程中哪些可提取物會真正遷移到藥品中。如果缺少這種相關(guān)性分析��,F(xiàn)DA可能質(zhì)疑研究對產(chǎn)品安全評估的價值�����。企業(yè)應(yīng)在提交資料中說明提取物與浸出物的對應(yīng)關(guān)系����,例如列出哪些提取出的化合物在成品中被檢測到,哪些未被檢測到及其原因����。
未提供浸出物隨時間變化的數(shù)據(jù): 沒有展示浸出物在貨架期內(nèi)隨時間的變化趨勢�����。FDA期望看到在加速和長期穩(wěn)定性條件下�����,浸出物濃度是否隨時間增加或保持穩(wěn)定。如果缺少這些趨勢數(shù)據(jù)����,監(jiān)管方無法評估浸出物的長期累積風(fēng)險。企業(yè)應(yīng)在穩(wěn)定性研究中監(jiān)測浸出物的含量變化��,并在提交資料中提供圖表或表格說明其隨時間的變化情況����,以證明浸出物在有效期內(nèi)不會顯著增加。
未提供參考對照數(shù)據(jù): 缺乏與對照樣品或參比制劑(RLD)的比較數(shù)據(jù)��。例如�����,在穩(wěn)定性研究中未設(shè)置未包裝的空白對照����,導(dǎo)致無法區(qū)分藥品本身的雜質(zhì)與包裝浸出物。又如�����,未將新申請產(chǎn)品的浸出物譜與原研產(chǎn)品進(jìn)行比較,從而無法證明差異在可接受范圍內(nèi)����。FDA可能要求企業(yè)補充對照樣品數(shù)據(jù),以確認(rèn)浸出物確實來自包裝系統(tǒng)而非藥品自身����。如果發(fā)現(xiàn)浸出物水平與原研產(chǎn)品不一致,需要進(jìn)一步解釋其合理性�����。
數(shù)據(jù)合理性缺乏充分論證: 對檢測結(jié)果的安全性解釋不充分����,未提供足夠的科學(xué)依據(jù)。例如����,某浸出物的含量雖然低于安全閾值,但未說明其毒性風(fēng)險極低或有其他措施控制其進(jìn)一步降低�����。FDA可能要求企業(yè)提供毒理學(xué)數(shù)據(jù)或其他支持����,證明即使浸出物水平未超過閾值,其對患者的風(fēng)險也是可接受的����。企業(yè)應(yīng)在提交資料中對每項浸出物的安全評估進(jìn)行詳細(xì)說明,包括毒性閾值����、每日攝入量、安全系數(shù)等�����,確保監(jiān)管方能夠理解數(shù)據(jù)的合理性��。
示例解讀: 某生物制品申請在審評中��,F(xiàn)DA發(fā)現(xiàn)提交資料中沒有將提取物分析與浸出物結(jié)果進(jìn)行關(guān)聯(lián)�����,也沒有提供浸出物隨時間變化的數(shù)據(jù)�����。FDA據(jù)此要求申請人補充分析,將提取出的化合物與成品中的浸出物對應(yīng)起來����,并提供貨架期內(nèi)浸出物濃度的變化趨勢。申請人補充分析后發(fā)現(xiàn)��,一些在提取中檢出的化合物并未在成品中出現(xiàn)�����,原因是配方中的某些成分與其發(fā)生了反應(yīng)��。這一發(fā)現(xiàn)雖然是預(yù)期之外的��,但申請人及時在回復(fù)中解釋了原因����,并指出這些化合物由于反應(yīng)而無法遷移到成品中,因此對安全無影響��。FDA接受了申請人的解釋�����,并認(rèn)可了其關(guān)聯(lián)分析和趨勢數(shù)據(jù)的補充,認(rèn)為研究設(shè)計得到了完善�����。
3. 法規(guī)合規(guī)性方面的不足
此類問題涉及E&L研究是否符合FDA的法規(guī)要求和指導(dǎo)原則��。FDA依據(jù)聯(lián)邦法規(guī)和指南來評估E&L研究的完整性和合規(guī)性����,若發(fā)現(xiàn)不符合之處將要求補充信息�����。以下是常見的法規(guī)合規(guī)類不足:
未遵守USP或ICH指南: 研究未遵循美國藥典(USP)或國際人用藥品注冊技術(shù)協(xié)調(diào)會(ICH)關(guān)于E&L評估的推薦方法�����。例如��,F(xiàn)DA在2021年Generic Drugs Forum會議上指出��,E&L研究應(yīng)遵循USP <1663>和<1664>的推薦��。如果企業(yè)未按照這些指南進(jìn)行研究設(shè)計和數(shù)據(jù)報告��,F(xiàn)DA可能要求調(diào)整研究以符合指南要求。此外����,ICH正在制定新的Q3E指南以統(tǒng)一E&L評估標(biāo)準(zhǔn)。在該指南發(fā)布后�����,F(xiàn)DA可能會將其納入審評標(biāo)準(zhǔn)�����。企業(yè)應(yīng)密切關(guān)注最新法規(guī)動態(tài)��,確保研究符合當(dāng)前適用的指南�����。
缺少必要的毒理學(xué)評估: 未對超過安全閾值的浸出物進(jìn)行充分的毒理學(xué)評估����。FDA要求對所有超過安全閾值的浸出物進(jìn)行毒性風(fēng)險評估,以確定其對患者的可接受性����。如果提交資料中缺少對某高濃度浸出物的毒理學(xué)分析(如未能提供其NOAEL或安全暴露水平)����,F(xiàn)DA可能要求補充��。例如�����,某浸出物的每日攝入量超過了ICH M7對潛在遺傳毒性雜質(zhì)的關(guān)注閾值��,而企業(yè)未進(jìn)行遺傳毒性研究或控制策略����,F(xiàn)DA會認(rèn)為這是嚴(yán)重的合規(guī)不足�����。企業(yè)應(yīng)確保對所有關(guān)鍵浸出物進(jìn)行毒理學(xué)評估�����,并在需要時提供相應(yīng)的試驗數(shù)據(jù)或文獻(xiàn)資料��,以證明其安全性�����。
缺少材料來源和變更控制信息: 未提供包裝材料供應(yīng)商的相關(guān)信息或變更控制計劃。FDA要求在申請中提供包裝材料的化學(xué)組成�����、制造工藝以及供應(yīng)商的DMF信息�����,以便監(jiān)管方審核�����。如果缺少這些信息��,F(xiàn)DA可能要求補充供應(yīng)商的DMF引用或材料規(guī)格說明����。此外,對于包裝材料的變更��,企業(yè)應(yīng)按照21 CFR 314.70等法規(guī)要求及時通知FDA并提供變更后的E&;L研究數(shù)據(jù)��。如果企業(yè)未在申請中包含變更控制計劃或相關(guān)變更資料�����,F(xiàn)DA可能質(zhì)疑其合規(guī)性。企業(yè)應(yīng)在申報資料中附上包裝材料的DMF引用�����、材料規(guī)格和變更控制策略��,以滿足法規(guī)要求�����。
未滿足CGMP和QbD原則: 研究設(shè)計或數(shù)據(jù)管理不符合現(xiàn)行藥品生產(chǎn)質(zhì)量管理規(guī)范(CGMP)和質(zhì)量源于設(shè)計(QbD)原則�����。例如��,未建立包裝材料的質(zhì)量標(biāo)準(zhǔn)和驗收方法��,或未對分析數(shù)據(jù)進(jìn)行完整的記錄和偏差處理�����。又如����,未將E&L評估納入藥品開發(fā)的質(zhì)量風(fēng)險管理流程,缺乏對風(fēng)險的識別和控制措施����。FDA可能要求企業(yè)完善質(zhì)量體系,確保E&L研究過程符合CGMP要求��,所有數(shù)據(jù)和決策都有充分依據(jù)����。企業(yè)應(yīng)在申報資料中體現(xiàn)對包裝材料質(zhì)量的控制措施(如定期檢驗、變更控制)以及對風(fēng)險的評估結(jié)果�����,以證明研究的合規(guī)性和質(zhì)量可控性����。
示例解讀: 某新藥申請在審評中,F(xiàn)DA發(fā)現(xiàn)申請人未提供橡膠塞材料的DMF引用和化學(xué)成分信息�����,也未說明材料是否經(jīng)過適當(dāng)?shù)臏缇幚?���。FDA據(jù)此要求申請人補充這些信息�����,并提交橡膠塞材料的DMF文件或詳細(xì)說明��。申請人隨后提供了材料供應(yīng)商的DMF編號和材料規(guī)格�����,表明該橡膠塞材料已通過FDA認(rèn)可的生物相容性測試�����。FDA認(rèn)可了申請人的補充信息��,認(rèn)為其材料來源和處理符合法規(guī)要求。這一案例說明����,在E&L研究中提供完整的材料信息和變更控制計劃是合規(guī)的重要方面。
結(jié)語
美國FDA對提取物和浸出物相容性研究的審查要求日益嚴(yán)格����,企業(yè)在新藥或仿制藥注冊申請中必須充分重視E&L評估的完整性和科學(xué)性����。常見的發(fā)補意見涵蓋研究設(shè)計��、數(shù)據(jù)解釋和法規(guī)合規(guī)等多個方面��。通過提前規(guī)劃和執(zhí)行完善的E&L研究����,并在申報資料中清晰展示研究結(jié)果及其合理性,企業(yè)可以有效減少FDA提出補充信息的風(fēng)險��。
面對FDA的發(fā)補意見�����,企業(yè)應(yīng)保持積極溝通�����,按照FDA的要求及時補充必要的數(shù)據(jù)和說明��。例如����,在設(shè)計階段就與FDA進(jìn)行預(yù)溝通��,尋求其對E&L研究方案的指導(dǎo)�����,有助于確保研究符合監(jiān)管期望�����。在收到FDA意見后�����,企業(yè)應(yīng)組織專業(yè)團(tuán)隊深入分析問題����,制定詳細(xì)的回復(fù)方案����,包括補充實驗、數(shù)據(jù)補充和理由闡述等����。
最后,隨著ICH Q3E等新指南的發(fā)布��,F(xiàn)DA對E&;L評估的標(biāo)準(zhǔn)將更加統(tǒng)一和明確����。企業(yè)應(yīng)密切關(guān)注法規(guī)更新,持續(xù)優(yōu)化E&L研究方法和數(shù)據(jù)管理�����,以確保產(chǎn)品在整個生命周期內(nèi)的安全性和合規(guī)性��。通過科學(xué)嚴(yán)謹(jǐn)?shù)腅&L研究和充分的資料準(zhǔn)備��,企業(yè)能夠提高FDA審評的通過率�����,加速產(chǎn)品上市進(jìn)程����,同時保護(hù)患者免受包裝材料潛在風(fēng)險的影響。
參考資料
[1]ELSIE Database – ELSIE Consortium https://elsiedata.org/elsie-database/
[2](PDF) Strengthening Extractable & Leachable Study … https://www.researchgate.net/publication/387503864_Strengthening_Extractable_Leachable_Study_Submissions_Best_Practices_to_Avoid_Regulatory_Deficiencies
[3]Strengthening Extractable & Leachable Study … https://www.scirp.org/pdf/ajac20241512_32202357.pdf
[4]Q3E Guideline for Extractables and Leachables | FDA https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q3e-guideline-extractables-and-leachables
[5]Strengthening Extractable & Leachable Study … https://www.scirp.org/journal/paperinformation?paperid=138466
[6]Strengthening Extractable & Leachable Study Submissions: … https://www.scirp.org/pdf/ajac20241512_32202357.pdf
[7](PDF) Strengthening Extractable & Leachable … https://www.researchgate.net/publication/387503864_Strengthening_Extractable_Leachable_Study_Submissions_Best_Practices_to_Avoid_Regulatory_Deficiencies
[8]FDA對ANDA申報中E/L研究數(shù)據(jù)有什么要求��? - 知乎 https://zhuanlan.zhihu.com/p/693736312
[9]Q3E Guideline for Extractables and Leachables | FDA https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q3e-guideline-extractables-and-leachables
[10]Challenges (single use) - CASSS https://www.casss.org/docs/default-source/wcbp/2024-roundtable-notes/extractables-and-leachables---new-ich-q3e-guidance-common-practices-and-challenges.pdf