本文對近年廣東省藥品監(jiān)督管理局受理的注射劑包材變更資料進行回顧分析���。結(jié)合國家對注射劑包裝材料和容器的相關(guān)法規(guī)及指導(dǎo)原則,梳理變更包裝材料和容器材質(zhì)的相關(guān)要求�����,包括包材變更原因�、可比性研究、相容性研究�����、密封性研究、工藝驗證(含包裝工藝驗證)及穩(wěn)定性等方面�����,并探討存在的變更問題�,旨在督促藥品上市許可持有人和藥品包裝生產(chǎn)企業(yè)提高注射劑包裝材料和容器變更的風險意識,切實履行主體責任���,為包材變更的研究工作提供一定參考�。
Part.01前言
1國家對注射劑包材的法規(guī)要求
藥包材主要是指直接接觸藥品的包裝材料和容器�����,應(yīng)具有良好的保護性�����、相容性���、安全性�����、功能性及自身穩(wěn)定性�����,在藥品的包裝�、貯存�����、運輸和使用過程中保證藥品的安全有效和質(zhì)量���,實現(xiàn)給藥目的 [1]���。
國家食品藥品監(jiān)督管理局于 2004年 7 月 20 日發(fā)布并施行《直接接觸藥品的包裝材料和容器管理辦法》,內(nèi)容包括藥包材的標準�、藥包材的注冊、藥包材的再注冊�����、藥包材的補充申請�����、復(fù)審、監(jiān)督與檢查以及法律責任等 [2]�。該辦法于 2021 年 4 月 22 日宣布廢止。國家藥監(jiān)局于 2025 年 1 月 2 日發(fā)布�,2026 年 1 月 1 日正式實施的《藥品生產(chǎn)質(zhì)量管理規(guī)范》藥包材附錄,從質(zhì)量管理�、機構(gòu)與人員、廠房與設(shè)施�、設(shè)備、物料與產(chǎn)品�、確認與驗證、文件管理���、生產(chǎn)管理���、質(zhì)量控制與質(zhì)量保證、產(chǎn)品發(fā)運與召回�、合同管理等多個方面對藥包材生產(chǎn)企業(yè)進行了相關(guān)規(guī)定,旨在確保持續(xù)穩(wěn)定地生產(chǎn)出符合藥用要求和預(yù)定用途的藥包材 [3]�。除常規(guī)生產(chǎn)和質(zhì)量相關(guān)規(guī)定外,第十二章 合同管理規(guī)定藥包材生產(chǎn)企業(yè)應(yīng)當與藥品上市許可持有人簽訂質(zhì)量協(xié)議���;建立合同評審相關(guān)規(guī)程�����;接受并配合藥品上市許可持有人的審核工作���。
《中華人民共和國藥典》2025 年版在 2015 年版藥典首次收載藥包材通用要求和玻璃類藥包材 2 個指導(dǎo)原則、2020 年版藥典首次收載 16 個藥包材通用檢測方法的基礎(chǔ)上�,形成了“1 項通用指導(dǎo)原則 +4 項材質(zhì)指導(dǎo)原則 +58 項檢測方法”的主要框架 [4],具體內(nèi)容詳見表 1���。
國家食品藥品監(jiān)督管理局于 2012年 11 月 8 日發(fā)布的《國家食品藥品監(jiān)督管理局辦公室關(guān)于加強藥用玻璃包裝注射劑藥品監(jiān)督管理的通知》對注射劑的藥用玻璃包裝進行了相關(guān)規(guī)定�����,藥品生產(chǎn)企業(yè)應(yīng)根據(jù)藥品的特性選擇能保證藥品質(zhì)量的包裝材料 [5]���。對生物制品、偏酸偏堿及對 pH 敏感的注射劑�,應(yīng)選擇 121℃顆粒法耐水性為 1 級且內(nèi)表面耐水性為 HC1 級的藥用玻璃或其他適宜的包裝材料。除此之外�,該通知還規(guī)定相容性研究需符合《藥品包裝材料與藥物相容性試驗指導(dǎo)原則》
(YBB00142002)等相關(guān)技術(shù)指導(dǎo)原則的要求。
《藥品包裝材料與藥物相容性試驗指導(dǎo)原則》(YBB00142002-2015)對相容性試驗測試方法的建立�、相容性試驗的條件(光照試驗、加速試驗���、長期試驗�、特別要求、過程要求等)���、重點考察項目(包材���、原料藥及制劑相容性)進行了相關(guān)規(guī)定。其中���,注射劑的相容性重點考察項目包括外觀色澤�、含量�����、pH 值�、澄明度、有關(guān)物質(zhì)�、不溶性微粒、紫外吸收和膠塞的外觀���?!痘瘜W藥品注射劑與藥用玻璃包裝容器相容性研究技術(shù)指導(dǎo)原則(試行)》于 2015 年 7 月 28 日發(fā)布�,對相容性研究的考慮要點、相容性研究的主要內(nèi)容與分析方法���、試驗結(jié)果分析與安全性評估進行了規(guī)定 [6]�。指導(dǎo)原則中,注射劑與使用的藥用玻璃包裝容器相容性重點關(guān)注內(nèi)容包括:玻璃的類型���、玻璃的化學組成�、玻璃容器的生產(chǎn)工藝�����、規(guī)格大小�����、玻璃成型后的處理方式���;藥品和處方的性質(zhì),如藥液的 pH 值���、離子強度等�����;以及制劑生產(chǎn)過程中的清洗���、滅菌等工藝對玻璃容器的影響�����,如洗瓶階段的干熱滅菌工藝���、制劑冷凍干燥工藝、終端滅菌工藝等�。
1.2 注射劑中硼硅藥用玻璃發(fā)展現(xiàn)狀和趨勢
藥包材按材質(zhì)可分為玻璃類、橡膠類�����、塑料類���、金屬類�、陶瓷類和其他類(如紙�����、干燥劑)�,也可以由兩種或兩種以上的材料復(fù)合或組合而成(如復(fù)合膜、鋁塑組合蓋等)�。常用的藥品包裝用玻璃材料有硼硅玻璃和鈉鈣硅玻璃(后者亦簡稱為鈉鈣玻璃)兩大類 [1]���。硼硅玻璃可根據(jù)配方中硼含量、線熱膨脹系數(shù)�、耐水性能等細分為高硼硅玻璃、中硼硅玻璃和低硼硅玻璃�����。根據(jù)耐水性能或其他特征可分為不同級別的耐水玻璃���,如 I 類玻璃、II 類玻璃和 III 類玻璃���。藥品包裝用玻璃容器主要有玻璃輸液瓶�����、玻璃安瓿���、玻璃注射劑瓶、預(yù)灌封注射器用玻璃套筒���、筆式注射器用玻璃組件和玻璃藥瓶等產(chǎn)品�。
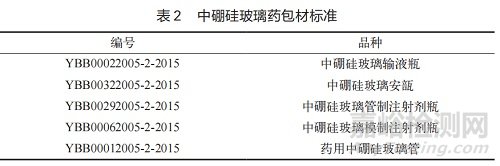
《藥用玻璃成分分類及理化參數(shù) 》(YBB00342003-2015)對高硼硅玻璃、中硼硅玻璃�、低硼硅玻璃和鈉鈣玻璃的化學組成及性能進行了規(guī)定。其中�����,中硼硅玻璃的成分標準為:B2O3 含 量 ≥ 8%�,SiO2 含量約為75%,Na2O+K2O 含量約為 4% ~ 8%���,MgO+CaO+BaO+SrO 含量約為 5%�,Al2O3 含量為 2% ~ 7%�����,平均線熱膨脹系數(shù)(20 ~ 300℃)為 3.5 ~ 6.1 10-6K-1�����,121℃顆粒耐水性為 1 級�����,98℃顆粒耐水性為 HGB1 級,耐酸性能為 1 級/100 μg/dm2���,耐堿性能為 2 級�����。國家藥包材標準(2015 年版)中對中硼硅玻璃的藥包材標準詳見表 2�。
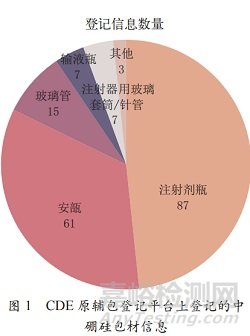
目前�����,在國家藥品監(jiān)督管理局藥品審評中心(CDE)的原輔包登記信息平臺上�,可查詢到中硼硅包材信息417 條,其中已激活的有 180 條�。在這180 條已激活的包材信息中,126 條為國產(chǎn)包材�,54 條為進口包材�����。其中有87 條為注射劑瓶�����,61 條為安瓿,15 條為玻璃管�,7 條為輸液瓶,7 條為注射器用玻璃套筒 / 針管�����,3 條為其他玻璃藥包材�,詳見圖 1。
1910 年�,德國肖特公司(Schott)發(fā)明了中硼硅玻璃,目前該公司占據(jù)全球約 50% 的中硼硅玻璃市場�,美國康寧公司(Corning)和日本電氣硝子株式會社(NEG)占據(jù)了全球約 30%的份額 [7]。由于在中硼硅玻璃的研發(fā)進度�����、生產(chǎn)技術(shù)和質(zhì)量標準體系等方面存在較大差異�����,我國中硼硅玻璃市場也主要由這三家公司壟斷供應(yīng)�。2020 年,國內(nèi)少數(shù)幾家企業(yè)只占據(jù)不到 13% 的市場份額 [8]�����。在相關(guān)政策的引領(lǐng)下,國產(chǎn)中硼硅包材正逐步進行更新?lián)Q代���,國內(nèi)部分藥品生產(chǎn)企業(yè)也開始使用國產(chǎn)中硼硅包材替代進口包材���。
Part.01內(nèi)包材變更的技術(shù)要求及常見問題分析
CDE 發(fā)布的《已上市生物制品藥學變更研究技術(shù)指導(dǎo)原則(試行)》《已上市化學藥品藥學變更研究技術(shù)指導(dǎo)原則(試行)》和《已上市中藥藥學變更研究技術(shù)指導(dǎo)原則(試行)》分別對生物制品、化藥和中藥的上市后變更作出了相關(guān)規(guī)定�,其中對于注射劑的內(nèi)包材材質(zhì)變更,分級均為重大變更���。將內(nèi)包材材質(zhì)由低硼硅變更為中硼硅�,根據(jù)前述通知 [5] 的要求�����,可以在省級藥品監(jiān)管部門進行備案�����,并按照重大變更的技術(shù)指導(dǎo)原則要求進行研究�,變更所需的申報資料主要包括以下幾類�����。
1包材的變更原因,變更后包材情況�,包材的選擇依據(jù)、合理性和適用性
企業(yè)需提供合理的包材變更原因�?����;幒椭兴幍淖兏笇?dǎo)原則中�����,只需要提供變更后的情況和質(zhì)量標準�����;生物制品的變更指導(dǎo)原則要求提供包材的選擇依據(jù)�、合理性和適用性,且要求不得使用國家禁止或淘汰的內(nèi)包材�。變更審評時應(yīng)關(guān)注企業(yè)提交的相關(guān)資料是否齊全,變更依據(jù)是否充分�。以下為典型問題示例:
包裝材料由“鈉鈣玻璃模制藥瓶和兒童安全阻開蓋(EVA 墊片)”變更為“口服液體藥用聚酯瓶(含配套瓶蓋)”,該品種適用人群包括兒童�����。依據(jù)藥審中心 2020 年發(fā)布的《兒童用藥(化學藥品)藥學開發(fā)指導(dǎo)原則(試行)》的要求,“包裝系統(tǒng)必須考慮將藥物包裝在防兒童開啟的容器內(nèi)�����,如兒童防護封閉設(shè)備 / 兒童安全蓋�����,以避免誤服�����。”建議企業(yè)在變更包材時�,同時考慮兒童給藥安全性要求�;
擬新增五層共擠輸液用膜的供應(yīng)商,企業(yè)提供變更前后包材的成分對比顯示各層包材組成成分不一致�;
企業(yè)未能提供藥用包材作為該品種包裝材料的批準證明性文件�。
2變更前后包材的對比信息及相關(guān)等同性 / 可替代性研究
化藥和中藥的變更指導(dǎo)原則要求企業(yè)進行包材的等同性 / 可替代性研究�����;生物制品的變更指導(dǎo)原則要求企業(yè)進行變更前后的對比�,如涉及相關(guān)內(nèi)容,應(yīng)提供包材的關(guān)聯(lián)審批信息�����。
除國家藥包材標準(YBB)外���,部分藥包材生產(chǎn)企業(yè)制定了內(nèi)部標準�����。在變更審評時需關(guān)注新舊包材的相關(guān)質(zhì)量標準是否存在差異�,原則上變更后包材質(zhì)量不應(yīng)低于變更前���。以下為典型問題示例:
企業(yè)未提供不同供應(yīng)商包材的特性對比研究及配方信息�;
未提供變更前后包材的質(zhì)量標準�,未結(jié)合新增供應(yīng)商的質(zhì)量標準制定相應(yīng)的包材內(nèi)控標準;
企業(yè)未能提供變更前后各一批包材的自檢報告(含關(guān)鍵質(zhì)量屬性)���,如透光率�����、內(nèi)表面耐水性及砷�����、銻���、鉛���、鎘的浸出量等。
3包材相容性研究�����、密閉完整性研究
指導(dǎo)原則規(guī)定�,中藥注射劑可根據(jù)品種情況進行包材相容性研究;化藥注射劑酌情進行包材相容性研究�����;生物制品需按照國內(nèi)外指導(dǎo)原則進行新包裝材料和容器與藥品的相容性研究�,以證明新包裝材料和容器對制劑質(zhì)量和安全性的影響�。指導(dǎo)原則規(guī)定,中藥和化藥的密封件應(yīng)開展包裝密封性研究���;生物制品需按照國內(nèi)外指導(dǎo)原則對密閉完整性進行研究�。
2.3.1 相容性研究
相容性研究一般應(yīng)包括以下幾部分內(nèi)容:
藥包材對藥物質(zhì)量影響的研究�����,包括藥包材的提取���、遷移研究及提取、遷移研究結(jié)果的毒理學評估�,藥物與藥包材之間發(fā)生反應(yīng)的可能性,藥物活性成分或功能性輔料被藥包材吸附或吸收的情況和內(nèi)容物的逸出及外來物的滲透等���;
藥物對藥包材影響的研究�����,考察經(jīng)包裝藥物后藥包材的完整性�、功能性及質(zhì)量的變化情況�,如玻璃容器的脫片、膠塞變形等�;
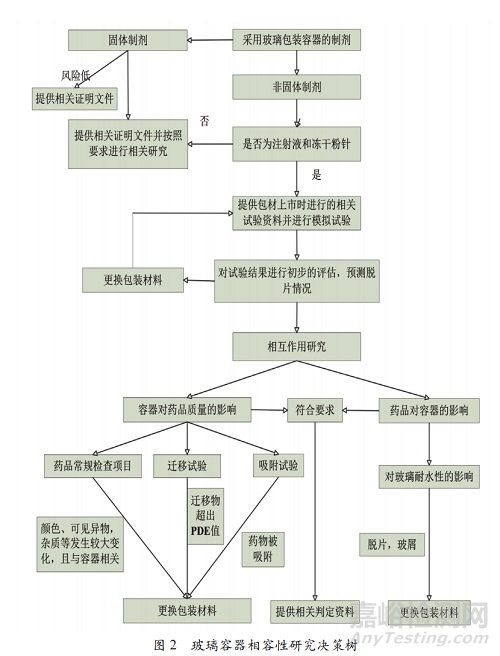
包裝制劑后藥物的質(zhì)量變化(穩(wěn)定性),包括加速試驗和長期試驗藥品質(zhì)量的變化情況���。對于存在多種包裝規(guī)格的注射劑應(yīng)選用比表面積最大的規(guī)格���。模擬溶劑的首選為對應(yīng)藥液���,若藥物對分析方法產(chǎn)生干擾,可經(jīng)評估選擇與制劑具有相同或相似理化性質(zhì)的模擬溶劑���。不同濃度的注射劑可參考 ICH 的括號法或矩陣法進行穩(wěn)定性研究�����。玻璃容器相容性研究決策樹見圖 2�����。
2.3.1.1 玻璃容器對藥品質(zhì)量的影響
玻璃容器對藥品質(zhì)量的影響包括對藥品常規(guī)檢查項目的影響�,即對藥品穩(wěn)定性的影響�,以及是否會引起可見異物、不溶性微粒���、重金屬含量超標等問題���。還包括對離子遷移的影響,需結(jié)合玻璃容器的組分以及添加物質(zhì)的信息進行檢查評估(常規(guī)金屬元素包括 Pb、Co���、Cd���、As、Li�、Sb、Ba�、Fe�����、Zn���、Cr 等)�,還需對提示玻璃被侵蝕或產(chǎn)生脫片趨勢的元素(如 Al�、B���、Si 等)進行檢查�。對于微量�����、治療窗窄、存在易于與玻璃發(fā)生吸附的官能團的藥物���,還需進行吸附試驗�����?��?蛇x擇加速試驗以及長期穩(wěn)定性試驗的考察時間點,按照藥品標準進行檢驗�����,并根據(jù)考察對象(如功能性輔料)適量增加檢驗項目�����。
2.3.1.2 藥品對玻璃容器的影響
藥品對玻璃容器的影響體現(xiàn)在玻璃容器被侵蝕后可能會出現(xiàn)脫片和微粒(玻屑)的現(xiàn)象�。通常存放時間越長,產(chǎn)生脫片的傾向越高���。因此可適當延長考察時間���,例如在加速條件下進行9 ~ 12 個月的考察���,以及在長期條件下進行相關(guān)評估。此外�����,也可以采用加速脫片的試驗方法�����。根據(jù)美國藥典(USP)<1660> 玻璃內(nèi)表面耐受性評估指南�,加速脫片的溶劑包括 pH8.0 的0.9%KCl 溶液���、pH8.0 的 3% 枸櫞酸鈉溶液和 pH10.0 的 20 mM 甘氨酸溶液�。在評估過程中���,可通過玻璃內(nèi)表面的侵蝕痕跡���、注射劑中的可見異物、注射液中的不溶性微粒以及 Si 元素或金屬離子的變化趨勢來進行綜合分析評估�����。
2.3.1.3 包裝制劑后藥物的質(zhì)量變化
需考察藥品在貯存、運輸及使用過程中可能面臨的最極端條件�����?��?蓞⒖挤€(wěn)定性試驗的時間點設(shè)置���,對注射液采用正立和倒置的方式分別進行試驗。
國內(nèi)部分藥品上市許可持有人不具備進行相容性研究的檢測能力�����,因此將對應(yīng)研究委托給相關(guān)檢測機構(gòu)�����。在審評時應(yīng)關(guān)注是否使用藥液進行相容性研究���,檢測機構(gòu)提供的報告是否研究充分�����,以及報告提交后委托方是否對報告進行了嚴格審核�。藥品的內(nèi)包材作為一個整體,在進行玻璃內(nèi)包材的變更相容性研究時�����,除了考察制劑與玻璃包裝容器的相容性外�,還應(yīng)關(guān)注制劑與其他組件及材料的相容性,如膠塞等也需進行相容性研究���。以下為典型問題示例:
未提供包材相容性研究資料�����;
提取液的選擇僅限于強酸和強堿提取�,未包含藥液提?��。?/span>
提取試驗選擇的溶劑和模擬條件�,未涵蓋樣品的 pH 范圍及實際生產(chǎn)工藝條件;
企業(yè)在開展包材相容性研究的同時�,也進行了元素雜質(zhì)研究,但未按照 ICH Q3D 的要求���,對注射劑必須研究的 1+2A 元素中 1 類元素 Cd���,Pb�����,Hg 建立相應(yīng)方法(含分析方法學驗證)并制定合理的控制策略���;
由于玻璃預(yù)灌封注射器的加工工藝導(dǎo)致鎢在玻璃套筒內(nèi)殘留,影響其內(nèi)表面耐受���,進而引發(fā)蛋白質(zhì)聚集或沉淀 [12]�����,因此應(yīng)控制預(yù)灌封注射器中鎢的溶出量���;
企業(yè)一次性申報了不同規(guī)格和濃度的產(chǎn)品變更包材供應(yīng)商,但僅對低濃度樣品開展了浸出物研究(0 月���、3 月���、6 月)�����,未對高濃度的樣品開展相應(yīng)的浸出物研究���。
2.3.2 密封性研究
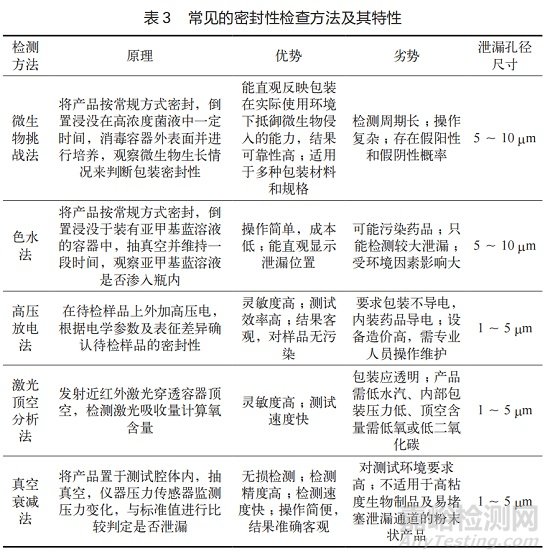
包裝系統(tǒng)密封性是指包裝系統(tǒng)防止內(nèi)容物損失、微生物侵入以及氣體或其他物質(zhì)進入���,保證藥品持續(xù)符合安全與質(zhì)量要求的能力 [9]�����?!吨袊幍洹吠▌t 0102 注射劑規(guī)定�����,容器的密封性須采用適宜的方法進行確證�����。注射劑包裝系統(tǒng)的泄漏類型包括微生物侵入�、藥品逸出或外部液體 / 固體的侵入以及頂空氣體含量的改變�����。常見的密封性檢查方法及其特性見表 3[10 -11]。
微生物挑戰(zhàn)法和色水法屬于概率性方法���,而高壓放電法���、激光頂空分析法和真空衰減法則屬于確定性方法。指導(dǎo)原則建議至少選擇兩種方法(其中一種推薦微生物挑戰(zhàn)法)進行密封性研究���,并對這兩種方法的靈敏度進行比較研究�。
在變更審評時應(yīng)關(guān)注企業(yè)是否綜合評估產(chǎn)品特性以確定密封性檢測方法���,以及該方法是否進行了適當?shù)姆椒▽W驗證�����。如產(chǎn)品有多個規(guī)格�,則每個規(guī)格都需要進行密封性研究���。以下為典型問題示例:
變更注射劑包材和容器供應(yīng)商�����、尺寸或形狀���,但未開展密封性研究�����;
包裝密封性研究未考慮變更后不同供應(yīng)商包裝組件的密封性���,若包材變更涉及多個生產(chǎn)場地,應(yīng)考慮不同場地包材密封性的研究 / 驗證�����;
變更產(chǎn)品的檢漏方法���,未提供密封性研究驗證資料或未對不同檢漏方法的靈敏度進行比較���;
檢漏工序中色水溶液由“0.05%亞甲基藍”變更為“0.03% 胭脂紅”,但未對變更前后的色水檢漏法進行平行驗證�。
4生產(chǎn)工藝驗證、過程控制
中藥和化藥注射劑的變更指導(dǎo)原則僅要求進行包裝工藝驗證���,若為無菌制劑�,必要時還需進行無菌 / 滅菌工藝驗證�����。生物制品變更指導(dǎo)原則要求提供變更后連續(xù)生產(chǎn)三批制劑的生產(chǎn)工藝和過程控制資料���,并與變更前進行對比�����;必要時還需進行產(chǎn)品的擴展性質(zhì)量研究���。
無菌制劑的無菌 / 滅菌工藝驗證需經(jīng)評估選擇適宜的批量,通常批量應(yīng)與生產(chǎn)批量相同�。產(chǎn)品的所有規(guī)格都應(yīng)進行無菌 / 滅菌工藝驗證。除無菌 /滅菌工藝驗證外���,還需關(guān)注濕熱滅菌 /干熱滅菌所涉及的相關(guān)設(shè)備的確認 / 驗證結(jié)果���,以及培養(yǎng)基模擬灌裝的方案和報告。這些應(yīng)與生產(chǎn)實際相符�����,且符合相關(guān)法規(guī)要求。以下為典型問題示例:
工藝驗證報告的滅菌工藝“121℃12min F0 ≥ 12”與批準文件載明的滅菌工藝“121℃ 15min F0 ≥ 12”不一致�;滅菌工藝驗證未按照工藝規(guī)程規(guī)定的溫度和時間進行研究,申請人未能提供相關(guān)滅菌工藝溫度變更的證明文件���,因此不認可其滅菌驗證參數(shù)�;
未對產(chǎn)品進行包裝工藝驗證�����,注射劑的包裝工藝未按照商業(yè)化批量進行驗證�����;
工藝驗證過程中���,其中一批采用了延長滅菌時間的方式進行糾正�,企業(yè)未能提供合理解釋�;
未提供無菌工藝驗證資料。
5變更后連續(xù)三批產(chǎn)品的批檢驗報告
檢驗結(jié)果應(yīng)符合質(zhì)量標準的規(guī)定���。如果經(jīng)評估需發(fā)起注冊檢驗���,注冊檢驗的結(jié)果也應(yīng)符合質(zhì)量標準的規(guī)定�����,且與企業(yè)自檢結(jié)果無較大差異。指導(dǎo)原則僅要求提供批檢驗報告�����,但在實際變更審評過程中�����,還需同步提供涉及的三批產(chǎn)品的生產(chǎn)信息���,如規(guī)格�����、批號�、批量等�����。以下為典型問題示例:
變更后產(chǎn)品的檢驗報告未包含產(chǎn)品批量信息,部分檢驗結(jié)果如過氧化值�、不溶性微粒、有關(guān)物質(zhì)等僅標注符合規(guī)定�����,未給出實際檢測結(jié)果�;
自檢報告與穩(wěn)定性總結(jié)報告數(shù)據(jù)不一致;
部分檢驗結(jié)果在省所自檢報告與企業(yè)自檢報告之間存在較大差異���,企業(yè)未能給出合理解釋�。
6穩(wěn)定性研究資料
中藥指導(dǎo)原則僅要求提供穩(wěn)定性研究資料并與變更前的穩(wěn)定性情況進行對比�����;化藥指導(dǎo)原則要求對三批樣品進行加速和長期穩(wěn)定性研究�����,申請時需提供 3 ~ 6 個月的穩(wěn)定性資料并與變更前的穩(wěn)定性情況進行對比���,確保變更后的穩(wěn)定性不低于變更前�,酌情進行使用中的穩(wěn)定性研究�����;生物制品指導(dǎo)原則要求提供商業(yè)化規(guī)模生產(chǎn)的新包裝制劑至少 3 個月的加速 / 強制降解穩(wěn)定性數(shù)據(jù)和至少 3 ~ 6 個月的長期穩(wěn)定性數(shù)據(jù),并與變更前的穩(wěn)定性情況進行對比���。同時需制定穩(wěn)定性方案�,繼續(xù)進行穩(wěn)定性研究�����,并承諾及時報告長期穩(wěn)定性研究中出現(xiàn)的任何不合格情況�����。
根據(jù)化學藥品注射劑與藥用玻璃包裝容器相容性研究指導(dǎo)原則(試行)[6]�,由于不同生產(chǎn)工藝對玻璃制品質(zhì)量的影響不同�,模制玻璃容器內(nèi)表面的耐受性基本相容,而管制玻璃由于工藝不同�����,其內(nèi)表面的耐受性有所差異�����。為了盡可能保證溶液與玻璃容器底部應(yīng)力環(huán)部位與肩部接觸,注射劑在穩(wěn)定性考察期間應(yīng)分別采用正立和倒置的方式進行試驗���。
根據(jù)《中國藥典》9402 生物制品穩(wěn)定性試驗指導(dǎo)原則的相關(guān)規(guī)定�����,生物制品液體制劑應(yīng)考慮與密閉系統(tǒng)的相互作用�����,樣品應(yīng)以倒立放置或水平放置�、正立放置兩種情況進行穩(wěn)定性試驗�����,原則上液體制劑應(yīng)與密閉系統(tǒng)充分接觸�����。
產(chǎn)品穩(wěn)定性可與包材相容性試驗同步進行�,方案設(shè)計時應(yīng)考慮對應(yīng)的考察時間點、考察項目和檢驗用量等因素�,選取足夠數(shù)量的產(chǎn)品進行穩(wěn)定性研究,以節(jié)省時間和人力成本�。
考察項目應(yīng)全面且具有代表性�,原則上關(guān)鍵時間點應(yīng)進行全檢�。變更后的穩(wěn)定性數(shù)據(jù)應(yīng)與變更前的穩(wěn)定性數(shù)據(jù)進行對比,含量�、雜質(zhì)等的變化應(yīng)能證明變更后產(chǎn)品質(zhì)量與變更前相當或更優(yōu)。以下為典型問題示例:
穩(wěn)定性試驗未采取倒置的方式考察粉末分裝工藝的樣品與新增供應(yīng)商膠塞的相互作用情況�;
未提供變更前后穩(wěn)定性數(shù)據(jù)的對比研究資料,應(yīng)提供變更后至少加速 6個月和長期 6 個月的穩(wěn)定性(含浸出物)考察數(shù)據(jù)�����;
未按照產(chǎn)品實際貯存條件開展穩(wěn)定性試驗考察���,若產(chǎn)品為常溫保存,建議長期試驗在 30±2℃�、65%±5%RH的環(huán)境下開展穩(wěn)定性研究;
不同規(guī)格樣品采用相同材質(zhì)(裝量一致)的包裝形式���,選擇最差工藝條件對其中一個規(guī)格進行 3 批工藝驗證���,但未提供其他規(guī)格樣品的穩(wěn)定性試驗數(shù)據(jù);
穩(wěn)定性研究數(shù)據(jù)顯示�,變更后加速 6 個月雜質(zhì)總量數(shù)據(jù)增長幅度明顯高于變更前,變更后樣品穩(wěn)定性低于變更前���,企業(yè)未能給出合理解釋�;
變更后穩(wěn)定性考察過程中不溶性微粒增長趨勢較變更前明顯,未結(jié)合包材相容性試驗結(jié)果進行合理分析�。
7其他技術(shù)要求
修訂質(zhì)量標準、說明書及標簽等信息�。根據(jù)生物制品指導(dǎo)原則的要求,若適用���,必須進行特殊安全性試驗(如過敏�����、溶血和血管刺激等)���。
Part.03小結(jié)與建議
隨著藥用包裝材料監(jiān)管法規(guī)的不斷健全與質(zhì)量標準體系的不斷完善,我國藥用玻璃的質(zhì)量正在不斷提高���。中性硼硅玻璃與藥物相容性研究為高質(zhì)量藥用玻璃包裝材料的使用做好了充足的準備�����,為新藥研發(fā)與包裝材料的選擇提供了參考 [13]�。
藥包材從單獨申請注冊證變更為藥品關(guān)聯(lián)審評,從剝離狀態(tài)變?yōu)榕c原輔料地位一致�,成為藥品的一部分,凸顯了它對于藥品質(zhì)量安全的重要性�����。藥品上市許可持有人和藥包材生產(chǎn)企業(yè)應(yīng)重視包材的質(zhì)量���,提高制劑風險防控意識�����,做好包材變更的研究驗證相關(guān)工作�。
1完善質(zhì)量管理體系���,確保產(chǎn)品持續(xù)合規(guī)
國家藥監(jiān)局于 2025 年發(fā)布了《藥品生產(chǎn)質(zhì)量管理規(guī)范(2010 年修訂)》藥用輔料附錄���、藥包材附錄公告�,并將于 2026 年 1 月 1 日實施。藥用輔料和藥包材生產(chǎn)企業(yè)應(yīng)及時改進設(shè)施設(shè)備并完善質(zhì)量管理體系���,確保符合藥用輔料附錄和藥包材附錄的各項要求�����。此外�,國家藥監(jiān)局于 2025 年 11 月 21日發(fā)布了《藥包材附錄檢查指導(dǎo)原則》的通知,省級監(jiān)督管理部門應(yīng)加強對包材生產(chǎn)企業(yè)的監(jiān)督管理�,藥包材生產(chǎn)企業(yè)應(yīng)保證產(chǎn)品持續(xù)合規(guī)。隨著藥包材的質(zhì)量要求及監(jiān)管要求的不斷提升�����,藥品持有人和藥包材生產(chǎn)企業(yè)應(yīng)落實持有人主體責任���,不斷完善質(zhì)量管理體系�����,加強產(chǎn)品質(zhì)量建設(shè)���。核心內(nèi)包材(如特殊規(guī)格西林瓶、生物制劑用膜材)供應(yīng)商集中度較高���,部分企業(yè)為降低成本選擇“非頭部供應(yīng)商”�,但對其生產(chǎn)過程(如原料純度�����、潔凈度控制)的審計頻次不足,易導(dǎo)致批次質(zhì)量波動�。基于產(chǎn)品質(zhì)量源于設(shè)計的理念�����,制劑企業(yè)應(yīng)加強對包材供應(yīng)商的審計�,謹慎變更包材,并對變更后的包材進行充分篩選研究�����,確保產(chǎn)品的關(guān)鍵質(zhì)量屬性在有效期內(nèi)符合質(zhì)量要求���。
2強化法規(guī)及指導(dǎo)原則的學習���,筑牢產(chǎn)品安全防線
在藥品包材變更審評中,相容性研究是確保安全的核心環(huán)節(jié)�。然而,部分企業(yè)對此的關(guān)注仍局限于“藥品注冊階段”���,忽視了產(chǎn)品上市后因處方微調(diào)、儲存條件變更或包材供應(yīng)商工藝升級而觸發(fā)的相容性再評估。這種“靜態(tài)合規(guī)”的思維���,可能導(dǎo)致溶出物超標或有效成分吸附等潛在風險���。隨著國家藥監(jiān)局藥審中心(CDE)就 ICH《Q3E :可提取物與浸出物指導(dǎo)原則》草案征求意見,相關(guān)評估體系正日趨完善���。為此�,藥品上市許可持有人必須樹立全生命周期管理理念���,主動跟蹤指南更新�����,嚴格依據(jù)現(xiàn)行規(guī)范設(shè)計與開展相容性研究�����,并精準撰寫申報資料���。這不僅是確保包材變更申報合規(guī)、高效推進的保障�����,更是從根本上規(guī)避藥品安全風險的必然要求。
參考文獻
[1] 國家藥典委員會. 中華人民共和國藥典:四部[M]. 2025年版. 北京:中國醫(yī)藥科技出版社�,2025.
[2] 國家食品藥品監(jiān)督管理局.直接接觸藥品的包裝材料和容器管理辦法:國家食品藥品監(jiān)督管理局令第13號[EB/OL].(2004-07-20)[2025-08-01].https://www.nmpa.gov.cn/yaopin/ypfgwj/ypfgbmgzh/20040720010101543.html.
[3] 國家藥品監(jiān)督管理局. 國家藥監(jiān)局關(guān)于發(fā)布《藥品生產(chǎn)質(zhì)量管理規(guī)范( 2010 年修訂)》藥用輔料附錄、藥包材附錄的公告 [EB/OL]. ( 2025-01-02)[2025-08-01].
https://www.nmpa.gov.cn /xxgk/ggtg/ypggtg/ypqtggtg/20250102142249169.html.
[4] 陳蕾�,俞輝,王彥���,等. 2025年版《中國藥典》藥包材標準體系概述[J]. 中國藥品標準���,2025,26(01):67-76. DOI:10.19778/j.chp.2025.01.010.
[5] 國家食品藥品監(jiān)督管理局. 國家食品藥品監(jiān)督管理局辦公室關(guān)于加強藥用玻璃包裝注射劑藥品監(jiān)督管理的通知[EB/OL].(2012-11-08)[2025-08-01].https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjyp/20121108120001728.html.
[6] 國家食品藥品監(jiān)督管理局. 國家食品藥品監(jiān)督管理總局關(guān)于發(fā)布化學藥品注射劑與藥用玻璃包裝容器相容性研究技術(shù)指導(dǎo)原則(試行)的通告[EB/OL].(2015-07-28)[2025-08-01]. https://www.nmpa.gov.cn/xxgk/ggtg/ypggtg/ypqtggtg/20150728120001551.html.
[7] 李朝圓�,李秀英,賀一峰�,等. 醫(yī)藥包裝用玻璃材料的研究進展[J]. 陶瓷學報,2023���,44(06):1103-1115. DOI:10.13957/j.cnki.tcxb.2023.06.005.
[8] 蔡冬雪�,匡波�����,毛露路�����,等. 中硼硅藥用玻璃發(fā)展現(xiàn)狀和趨勢[J]. 玻璃�����,2023�����,50(01):20-29.
[9] 國家藥品監(jiān)督管理局藥品審評中心. 國家藥監(jiān)局藥審中心關(guān)于發(fā)布《化學藥品注射劑包裝系統(tǒng)密封性研究技術(shù)指南(試行)》和《化學藥品注射劑生產(chǎn)所用的塑料組件系統(tǒng)相容性研究技術(shù)指南(試行)》的通告(2020年第33號)[EB/OL].(2020-10-21)[2025-08-01]. https://www.cde.org.cn/main/news/viewInfoCommon/8a4f9f16844fbed617f8e8ed59485c1d.
[10] 付沛林�,王明建,王亮. 藥品密封性檢測:用戶需求與優(yōu)化[J]. 流程工業(yè)���,2025(04):36-40.
[11] 封二飛���,張筱宜,趙明. 無菌藥品容器密封完整性檢測方法對比研究[J]. 中國藥業(yè)���,2021�����,30(14):51-54.
[12] 劉春月���,張曼茹�����,秦洋�,等. 預(yù)灌封注射器鎢溶出量測定法制訂與解析[J]. 醫(yī)藥導(dǎo)報���,2025�,44(07):1055-1056.
[13] 陳婧�����,等. 中性硼硅玻璃與藥物相容性研究進展[J]. 包裝工程���,2021�,42(10):41.