20多年前���,美國出臺相關立法推動兒科藥物研發(fā)��,同時也引發(fā)了對藥物對兒科發(fā)育系統(tǒng)毒性的關注�����。這些立法包括2002年的《兒童最佳藥物法案》(BPCA)和2003年的《兒科研究公平法案》(PREA)��。當時的臨床前動物試驗主要針對成年動物���,在發(fā)育中的動物身上進行安全性評價尚屬新興領域��。人們期望通過幼齡動物研究(JAS)��,解答藥物在發(fā)育中動物體內潛在毒性的相關問題���。
2006年,FDA發(fā)布《兒科藥物產品非臨床安全性評價》行業(yè)指南�����,試圖使JAS標準化�����,并以特定毒理學問題為導向。該指南強調��,開展JAS應基于對動物和成人毒性數據審查后發(fā)現(xiàn)的不足���,圍繞具體問題展開��。
早期對藥物研發(fā)中JAS的評估發(fā)現(xiàn)��,在400種產品中���,僅有不到10%的處方信息(PIs)包含JAS相關內容。2007年�����,《FDA修正案法案》對BPCA和PREA進行修訂���,要求若開展了JAS,需將研究數據納入處方信息���。
JAS對兒科藥物研發(fā)成果的適用性存在不確定性��。2013年���,Baldrick指出���,JAS中發(fā)現(xiàn)的、有別于常規(guī)成年動物試驗的獨特結果十分罕見���,且“減少動物試驗”是未來藥物研發(fā)測試的趨勢��。僅用于證實成年動物中已發(fā)現(xiàn)毒性的JAS并無實際價值��。
2021年�����,ICH與FDA聯(lián)合發(fā)布了支持兒科藥物非臨床測試的新指南�����,該指南更側重于提供一種“證據權重法”(WoE)��,用于指導藥物研發(fā)過程中是否開展JAS的決策��。
3Rs原則(替代��、優(yōu)化���、減少)指在監(jiān)管安全性評估中�����,將重點從動物毒性研究轉向非動物測試���,即“新方法學”(NAMs,亦稱為“新型替代方法學”)�����。該原則已被美國FDA�����、美國國立衛(wèi)生研究院及其他聯(lián)邦機構廣泛采納�����。
盡管JAS被推薦作為兒科臨床前安全性評估的一部分���,但目前尚無關于這些研究對兒科藥物研發(fā)成果影響的最新評估。FDA做了一項研究,旨在實現(xiàn)兩大目標:1)從兒科產品的處方信息(PIs)中識別���、提取并分析JAS研究��;2)結合最新指導原則��,評估這些JAS研究得出的結果���。
FDA通過標簽檢索初步獲得269項結果,經交叉核查藥物是否受BPCA���、PREA法案或兩者共同推動��,并剔除重復項�����、505(B)(2)類申請藥物及未獲批用于兒科人群的藥物后��,最終確定74種獨特的兒科獲批產品�����。此外���,另有48種無獲批兒科適應癥的產品被單獨分析�����。
時間分布
74種產品中�����,含JAS的PIs數量隨年份呈增長趨勢:1955-1978年:僅6份�����;1979-2002年(BPCA通過前后):17份�����;兒科相關立法通過后�����,含JAS的PIs數量在近20年保持穩(wěn)定:2003-2012年為23份���,2013-2023年為28份���。
治療領域分布
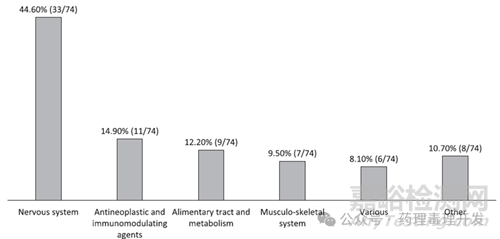
74種產品涉及多個治療領域(如下圖)��,其中JAS研究占比最高的領域為:神經系統(tǒng)藥物:44.6%(33/74)>抗腫瘤與免疫調節(jié)劑:14.9%(11/74)>消化道與代謝藥物:12.2%(9/74)>肌肉骨骼系統(tǒng)藥物:9.5%(7/74)���;還有些其它類別���,占比 10.7%(8/74),涵蓋全身用抗感染藥���、心血管系統(tǒng)藥物�����、血液及造血器官藥物���、皮膚病藥物、全身用激素類制劑等���。
JAS試驗種屬選擇
依據已發(fā)布指導原則�����,JAS應盡可能采用單一物種開展�����。74種產品的JAS物種選擇情況為:
83.8%(62/74)的產品采用單一物種進行JAS���;
16.2%(12/74)的產品采用2個物種���;
89.2%(66/74)的JAS以大鼠為實驗模型,為最常用物種�����;其次為小鼠(3項)��、犬(2項)��、小型豬與猴(各3項)�����。
基于“證據權重法”(WoE)標準的特征分析
結合指導原則中“目標患者最小年齡”和“臨床治療持續(xù)時間”兩項WoE客觀標準��,74種產品的特征如下:
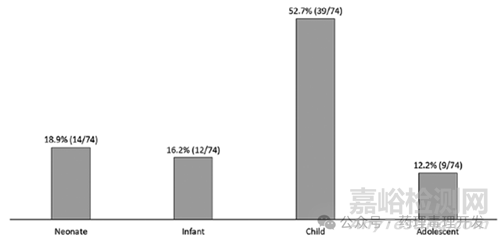
目標患者最小年齡(如下圖):1)針對低齡兒科患者(新生兒�����、嬰兒):35.1%(26/74);2)針對兒童與青少年:
64.9%(48/74)��?����?梢钥闯龃蟛糠諮AS研究是為了支持兒童這個階段的用藥�����。
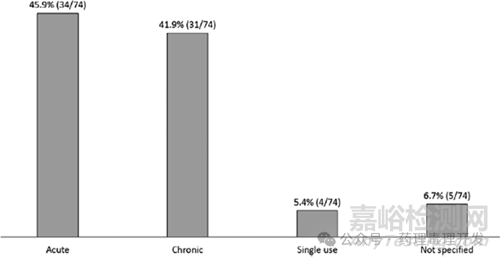
臨床治療持續(xù)時間(如下圖):1)慢性用藥(≥3個月):41.9%(31/74)�����;2)急性或單次用藥:51.4%(38/74)���。短期用藥稍微偏多一些。
JAS結果對藥品標簽的影響
兒科獲批產品:74種產品中�����,僅8.1%(6/74)的JAS數據直接影響標簽內容�����,促成了針對兒科人群的特定建議。例如:Aliskiren因幼年動物中轉運體和代謝酶未成熟導致藥物暴露量過高��,標簽注明“2歲以下患者禁用”�����;Linaclotide黑框警告提示“2歲以下患者禁用”���,因幼年小鼠給藥后出現(xiàn)脫水死亡��。如前文所言���,JAS中發(fā)現(xiàn)的、有別于常規(guī)成年動物試驗的獨特結果十分罕見���。
無兒科獲批適應癥的產品:48種產品中�����,僅8.3%(4/48)的JAS結果直接影響標簽�����。例如plecanatide的黑框警告注明“6歲以下患者禁用”���,因幼年小鼠給藥后出現(xiàn)脫水死亡���。
討論
JAS的現(xiàn)狀與特征
核心地位與數據概況:JAS是當前兒科臨床前發(fā)育安全性評估的核心。本研究發(fā)現(xiàn)���,美國FDA批準的藥品中,有74種在PI中提及JAS且
適用于兒科人群��。
數量變化趨勢:2002年兒科相關立法出臺后���,含JAS的藥品說明書數量發(fā)生大幅變化��,此后20年間保持穩(wěn)定��。
研究設計特點
物種選擇:多數研究僅采用單一物種��,以大鼠為主��。
適用人群:超半數藥品針對兒童和青少年���。
用藥場景:約半數藥品用于急性治療或單次使用。
對藥品標簽的影響:74種獲批兒科藥品中,僅6種(8.1%)的JAS結果直接影響了藥品標簽(如兒科用藥部分或警告項)��,這與對未獲批兒科藥品的分析結果(48種中有4種�����,占8.3%)相近���。
主要治療領域:神經作用類藥品的JAS開展數量最多�����。
兒科臨床前發(fā)育安全性評估的新方法探索
探索必要性:既響應FDA開發(fā)NAMs以減少動物實驗的目標���,更旨在提升兒科臨床前檢測結果對藥品標簽的直接影響。
潛在新方法如下:
拓展次要藥理學研究:制藥企業(yè)在IND申請階段對藥物次要靶點的檢測可用于篩查不良效應傾向�����,已有研究發(fā)現(xiàn)480家申辦方提交的1120項IND中���,對747種不同受體進行了結合試驗�����,此類分析已趨常規(guī)化���。
發(fā)育生物學受體研究:目前尚不清楚哪些發(fā)育生物學受體可預測兒科發(fā)育風險及747種已知受體中哪些對兒科患者有風險���;美國國立衛(wèi)生研究院(NIH)啟動的“發(fā)育基因型-組織表達(dGTEx)”項目,旨在建立資源數據庫和組織庫��,研究人和非人靈長類發(fā)育階段多參考組織的基因表達模式���,有望為篩選兒科發(fā)育相關受體提供支持。
微生理系統(tǒng)與器官芯片:已應用于多種疾病研究�����,可識別神經退行性疾病等的治療策略��。
定量構效關系(QSAR):已用于預測藥物誘導的肝損傷�����,在兒科發(fā)育安全性評估中潛力廣闊�����。
機器學習與人工智能(AI):2017年Boland等人開發(fā)的算法預測fetal loss和congenital abnormalities的準確率分別達91%和87%;Evangelista等人開發(fā)的Reprotox-KG平臺可識別與出生缺陷相關的生物通路�����,未來AI應用將進一步拓展�����。
建模與模擬:基于生理的藥代動力學(PBPK)模型可分析藥物暴露濃度��;定量系統(tǒng)藥理學(QSP)可增加模型的生物復雜性���,如某酸鞘磷脂酶缺乏癥(ASMD)的QSP模型結合臨床數據��,支持了奧利普酶α在兒科患者中的獲批��。
當前挑戰(zhàn)
2025年兒科安全性研究面臨困境��,兒科罕見病藥物研發(fā)處于高峰�����,但可參與長期安全性研究的兒科患者數量極少���;ICH E11增補文件將成人患者藥物安全性外推至兒科患者納入監(jiān)管指導���,這可能削弱20多年前推動JAS發(fā)展的核心考量,因此需將JAS與高效NAMs結合�����,實現(xiàn)全面的兒科臨床前發(fā)育安全性評估��。
引自:Pediatric Developmental Drug Toxicity:Description of Juvenile Animal Studies in US FDA Prescribing Information and Assessing the Need for New Approach Methodologies