1995年,Pre-IDE項(xiàng)目成立�����,該方式旨在為提交者(submitters)提供一種機(jī)制�,以便在未來的IDE申請(qǐng)?zhí)峤恢矮@得FDA的反饋���,隨著時(shí)間的推移�����,Pre-IDE項(xiàng)目發(fā)展到包括PMA/HDE/De Novo/510(K)提交的反饋���,以及解決臨床研究是否需要提交IDE。
2012年�����,F(xiàn)DA承諾建立一個(gè)結(jié)構(gòu)化的過程來管理這些提交者和FDA的反饋互動(dòng)�����,稱為“Pre-Submissions-預(yù)審核”���,并在2014年發(fā)布指南���,實(shí)施了更廣泛的Q-Submission (Q-Sub) 項(xiàng)目�����,其中包括了Pre-Submissions-預(yù)審核�,以及與FDA接觸的額外機(jī)會(huì)�。
在MDUFA III時(shí)期,行業(yè)和及機(jī)構(gòu)同意改進(jìn)Q-Sub計(jì)劃�,改變前期會(huì)議的安排,并修改FDA反饋時(shí)間的新績(jī)效目標(biāo)�。作為2022年醫(yī)療器械用戶費(fèi)用修正案(MDUFA V)的一部分,這些目標(biāo)得到了進(jìn)一步的細(xì)化���,該機(jī)構(gòu)還承諾發(fā)布一份指南更新草案�,其中包括更多信息�,以幫助提交人和審查人員確定提交人的問題最適合非正式溝通的情況,而不是預(yù)先提交���。
2023年6月2日�,F(xiàn)DA發(fā)布有關(guān)Q-sub的最新指南�,本次Q-sub指南更新的目的是在制定征求公眾意見的指南草案期間提供一些小的程序更新和澄清�。今天就為廣大制造商抽絲剝繭�,詳細(xì)介紹我們最常用的Pre-Sub預(yù)審核���。
1�、預(yù)審核Pre-Sub介紹
Pre-Sub包括提交者向FDA反饋的正式書面請(qǐng)求�,并以正式的書面回應(yīng)形式提供,如果提交者選擇�����,會(huì)議后的正式書面反饋���。在會(huì)議期間發(fā)生的所有討論都被記錄在由提交者起草并提交給FDA審查的會(huì)議記錄中���。
Pre-Sub為提交者提供了在預(yù)定上市前提交(即IDE、PMA�����、HDE�、De Novo請(qǐng)求、510(k)�、Dual�、BLA���、IND)等獲得FDA反饋的機(jī)會(huì)���。常見的問題有生物相容性、臺(tái)架測(cè)試�、網(wǎng)絡(luò)安全等。當(dāng)FDA對(duì)特定問題的反饋有助于指導(dǎo)產(chǎn)品開發(fā)和/或提交準(zhǔn)備時(shí)�,Pre-Sub是合適的,但FDA不打算對(duì)擬提交的文件進(jìn)行預(yù)審查或?qū)μ峤晃募刑峁┑臄?shù)據(jù)進(jìn)行預(yù)審查�。
預(yù)審核是提交者的自愿行為,預(yù)審核的好處在于提高后續(xù)項(xiàng)目提交�,縮短總審查時(shí)間,并促進(jìn)新器械的開發(fā)過程���。
FDA認(rèn)為���,在預(yù)審核中提供的互動(dòng)可能有助于FDA和提交者獲得更有效和透明的審查過程。CDRH和CBER的工作人員通過考慮多種符合最輕要求和原則的科學(xué)和監(jiān)管方法���,為前期制定反饋�����,以簡(jiǎn)化監(jiān)管流程���。FDA發(fā)現(xiàn)���,在執(zhí)行計(jì)劃的測(cè)試之前���,要求反饋是最有效的���。FDA在預(yù)審核中提出的問題并不要求提交者在隨后的提交中處理或解決這些問題,盡管任何與該主題相關(guān)的未來提交都應(yīng)討論為什么選擇不同的方法或未解決的問題���。此外���,對(duì)預(yù)審核中的信息進(jìn)行審查并不能保證在未來的提交中做出有利的決定。在對(duì)未來提交的文件進(jìn)行審查時(shí)�����,如果所有信息被視為一個(gè)整體���,或者在預(yù)審后出現(xiàn)了新的信息�,則可以提出其他問題。
在納入上市前提交之前�����,F(xiàn)DA還強(qiáng)烈建議使用預(yù)審核�����,以獲得對(duì)預(yù)定變更控制計(jì)劃(PCCPs)的反饋�。PCCP是根據(jù)FD&C法案第515C條添加的,允許制造商在PCCP獲得FDA授權(quán)后在PCCP范圍內(nèi)進(jìn)行修改�。
根據(jù)第515C條規(guī)定,F(xiàn)DA可以批準(zhǔn)或批準(zhǔn)PCCP用于描述可能對(duì)器械進(jìn)行的計(jì)劃更改的器械�����,否則需要補(bǔ)充上市前批準(zhǔn)申請(qǐng)或上市前通知�。例如,第515C條規(guī)定�����,如果器械變更與FDA批準(zhǔn)或批準(zhǔn)的PCCP一致�����,則不需要補(bǔ)充上市前批準(zhǔn)申請(qǐng)(第515C(a)條)或上市前通知(第515C(b)條)。第515C條還規(guī)定�����,F(xiàn)DA可以要求PCCP包括安全有效使用器械的標(biāo)簽�����,因?yàn)樵撈餍蹈鶕?jù)該計(jì)劃進(jìn)行了更改���,如果該器械不按照該計(jì)劃的預(yù)期功能進(jìn)行通知要求,以及根據(jù)計(jì)劃進(jìn)行更改的性能要求�。FDA鼓勵(lì)使用Pre-Sub,因?yàn)樗峁┝伺cFDA積極合作開發(fā)PCCP的機(jī)會(huì)���,這有助于簡(jiǎn)化上市前審查�。
2���、預(yù)審核時(shí)間表/推薦審核內(nèi)容和提交過程
2.1FDA預(yù)審核時(shí)間表
2.2推薦提交內(nèi)容
a.告知FDA提交者計(jì)劃的后續(xù)提交類型
FDA建議提交者明確指出未來的提交類型(比如510K/PMA/IDE/IND等)�����,明確后續(xù)提交類型是Pre-Sub問題的重點(diǎn)���,以幫助指導(dǎo)FDA的反饋�����。
b.提供背景資料
FDA建議提交者提供足夠的背景信息和支持文件�,以便FDA對(duì)提交者提出的Pre-Sub問題進(jìn)行反饋�。這些信息可能包括文獻(xiàn)文章、完整的設(shè)備描述和工程圖紙�、建議的標(biāo)簽、視頻和/或根據(jù)不同版本的協(xié)議修訂���,這取決于提交者要求反饋的具體問題�。包括提交者如何處理或計(jì)劃處理相關(guān)指導(dǎo)文件�����、法規(guī)���、特殊控制或其他適用于提交者的設(shè)備或提交類型的來源也可能會(huì)有所幫助�。
FDA也建議提交者提交完整背景資料很重要���,但要切題�,保證提交背景資料的針對(duì)性和重點(diǎn),要清楚的指出那些背景信息是和FDA討論的具體問題和主題相關(guān)的�����。提交無關(guān)的資料可能會(huì)起反作用���!
c.具體問題
預(yù)審核應(yīng)包括與計(jì)劃的后續(xù)提交類型相關(guān)的審查問題(例如�,關(guān)于非臨床和臨床測(cè)試方案或支持提交所需的數(shù)據(jù)的問題)�����,問題要明確���、具體,以便FDA和提交者將精力集中在與推進(jìn)項(xiàng)目最相關(guān)的問題上���。FDA建議提交者仔細(xì)考慮每個(gè)預(yù)審核中要求的主題數(shù)量和反饋范圍���,以確保FDA有足夠的時(shí)間在中對(duì)每個(gè)主題的問題進(jìn)行深度回應(yīng),并確保有針對(duì)性的會(huì)議�。
一般而言,F(xiàn)DA發(fā)現(xiàn)很難在一個(gè)預(yù)審核中解決超過3-4個(gè)實(shí)質(zhì)性主題。一個(gè)實(shí)質(zhì)性的主題涉及一個(gè)專注的專業(yè)領(lǐng)域�����。實(shí)質(zhì)性主題的例子包括但不限于臨床前/實(shí)驗(yàn)室測(cè)試�、生物相容性、動(dòng)物研究���、PCCP�����、軟件�、滅菌�����、臨床研究終點(diǎn)和統(tǒng)計(jì)分析計(jì)劃等�。
因此,F(xiàn)DA建議確定3-4個(gè)實(shí)質(zhì)性的主題���,因?yàn)檫@有助于召開更有效的會(huì)議�,并產(chǎn)生更有效的對(duì)話和反饋�����。其他的問題(例如,行政主題)可能是適當(dāng)?shù)?,如果他們可以解決,沒有深入的審查�,并且不引入新的重要主題。
如果提交者的問題包含過多的主題�����,F(xiàn)DA可能會(huì)與提交者聯(lián)系�,討論提交者希望優(yōu)先考慮哪些主題。在某些情況下�,F(xiàn)DA可能會(huì)建議在隨后的pre - sub中討論較低優(yōu)先級(jí)的主題。
以下是在預(yù)審中提交的常見類型問題的補(bǔ)充指引:
研究方案
資源限制不允許FDA準(zhǔn)備或設(shè)計(jì)特定的研究計(jì)劃�����。如果提交者希望獲得FDA對(duì)方案的反饋�����,他們應(yīng)該提交建議大綱�,并說明所選擇方法的合理性���。
為了獲得更有成效的反饋���,F(xiàn)DA建議提交者包括關(guān)于他們方案的具體問題�。如果沒有直接的問題�,F(xiàn)DA的反饋可能更加籠統(tǒng),很可能沒回答提交者感興趣的問題的足夠細(xì)節(jié)�。
如果預(yù)審核是用于非顯著風(fēng)險(xiǎn)器械研究、IDE豁免器械�����、CW���、Dual或提交者計(jì)劃在美國(guó)以外(OUS)進(jìn)行的研究���,以支持營(yíng)銷提交,提交者應(yīng)考慮在啟動(dòng)研究之前通過預(yù)審核提交整個(gè)方案�,特別是如果它提出了獨(dú)特的科學(xué)或監(jiān)管方面的考慮。
數(shù)據(jù)審核
預(yù)審核不適合數(shù)據(jù)審核項(xiàng)目�����。但如果數(shù)據(jù)和結(jié)論難以解釋���,可能應(yīng)就初步結(jié)果的解釋或計(jì)劃在即將提交的報(bào)告中處理結(jié)果的方法提出一個(gè)具體問題�。
監(jiān)管方法
在預(yù)審核下,F(xiàn)DA能夠提供關(guān)于監(jiān)管策略和方法的反饋�����。例如���,產(chǎn)品上市路徑是510K還是De Novo�����。當(dāng)然�����,器械分類和使用適應(yīng)癥的正式書面請(qǐng)求還需要需要進(jìn)行513(g)�,獲得FDA進(jìn)一步的回復(fù)�。
2.3審查過程
a.驗(yàn)收評(píng)審
在審查時(shí)鐘開始后的15天內(nèi),F(xiàn)DA工作人員將使用驗(yàn)收檢查表進(jìn)行驗(yàn)收審查���。
驗(yàn)收審查完成�����,提交者將收到關(guān)于提交是否已被接受進(jìn)行評(píng)審的通知�����,以及首席評(píng)審人員或RPM的聯(lián)系信息�。如果Pre-Sub要求召開會(huì)議被接受�,此通知也將確認(rèn)提交人要求的會(huì)議日期或提供兩個(gè)安排在第75天之前的替代會(huì)議日期。
如果驗(yàn)收評(píng)審確定申請(qǐng)不符合Pre-Sub要求或內(nèi)容提交不完整�,項(xiàng)目會(huì)被FDA拒絕受理(Refuse to Accept-RTA),F(xiàn)DA會(huì)給提交者發(fā)布RTA信函并解釋拒絕原因���。提交者可以通過向DCC提交額外信息來回應(yīng)RTA�����,這些信息將作為Q-Sub的修正案登錄���。
收到新提交的信息后,審查時(shí)鐘將在第0天重新啟動(dòng)���,F(xiàn)DA工作人員將在重新啟動(dòng)審查時(shí)鐘的前15天內(nèi)按照相同的程序再次進(jìn)行驗(yàn)收審查�����。隨后的驗(yàn)收審查將根據(jù)驗(yàn)收清單評(píng)估新信息是否使提交完成���。
b.會(huì)議安排
如果可行�����,F(xiàn)DA將在提交者要求的會(huì)議日期之一安排會(huì)議�。在FDA收到您的提交后的70-75天內(nèi)召開會(huì)議是最可行的���。如果FDA不能滿足提交者要求的其中一個(gè)日期���,F(xiàn)DA將提供至少兩個(gè)備選日期,這些日期在收到已接受提交的日期75天之前���。
FDA打算在收到已接受的提交后30天內(nèi)與提交人就會(huì)議日期達(dá)成協(xié)議�����。對(duì)于在收到已接受的提交材料后30天內(nèi)沒有商定會(huì)議日期的所有會(huì)議請(qǐng)求���,F(xiàn)DA manager將在第40天前聯(lián)系提交者解決日程安排問題。
c.反饋
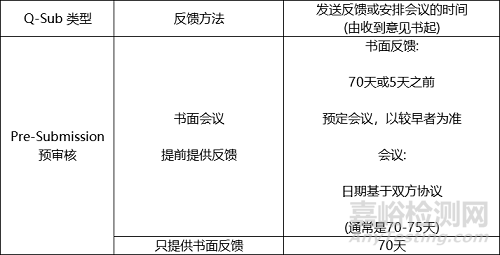
書面反饋將通過電子郵件提供給提交者,包括:對(duì)提交者問題的書面答復(fù);FDA對(duì)會(huì)議額外議題的建議(如適用);或者兩者都有�����。FDA打算遵循以下時(shí)間表向預(yù)審核提供反饋�。
預(yù)審核書面反饋:如果提交者沒有要求開會(huì)�����,將會(huì)有書面反饋�����,F(xiàn)DA在收到已接受的意見書后70個(gè)日歷日內(nèi)提供�,并將作為FDA相關(guān)部門反饋意見的正式記錄���。
預(yù)審核會(huì)議:如果要求召開會(huì)議�,將在預(yù)定會(huì)議前至少5天���,最遲在收到接受的預(yù)審核后70天提供書面反饋���。如果提交者的所有問題都得到了解決,提交者可以取消會(huì)議,書面答復(fù)將作為FDA相關(guān)部門反饋的正式記錄���。如果召開會(huì)議���,會(huì)議記錄與書面反饋一起將構(gòu)成FDA相關(guān)部門反饋的正式記錄。
在收到FDA書面反饋和召開會(huì)議之間或在會(huì)議期間�����,F(xiàn)DA不應(yīng)期望審查和回應(yīng)提交者準(zhǔn)備的并提供給FDA的額外信息�����,因?yàn)镕DA沒有足夠的時(shí)間對(duì)這些信息進(jìn)行徹底審查���。任何需要額外FDA審查的信息都應(yīng)作為Pre-Sub的補(bǔ)充或在最終上市前提交�。然而�����,適當(dāng)?shù)淖龇ㄊ强s小提交者的議程�,集中于反饋中的特定問題或主題。
FDA的反饋基于提交者提供的信息和當(dāng)時(shí)已知的當(dāng)時(shí)技術(shù)水平提出的最佳建議���。一般情況下�,F(xiàn)DA針對(duì)預(yù)審核提供的反饋不會(huì)改變,前提是后續(xù)提交者正式提交的信息和預(yù)審核提交的信息一致���,并且在正式提交時(shí)���,當(dāng)前最新技術(shù)水平與預(yù)審核時(shí)的一致�,不會(huì)對(duì)安全有效性產(chǎn)生重大影響。
FDA對(duì)反饋修改的情況僅限于:
1)出現(xiàn)新的科學(xué)發(fā)現(xiàn)�����,表明存在新的風(fēng)險(xiǎn)或已知風(fēng)險(xiǎn)的發(fā)生頻率增加���,影響FDA之前的建議,F(xiàn)DA可能會(huì)修改之前預(yù)審核的反饋;或者是否有新的公共衛(wèi)生問題影響FDA之前的建議�����。
2)該器械的預(yù)期用途�、設(shè)備技術(shù)或標(biāo)簽進(jìn)行了重大更改�,或提供有關(guān)設(shè)備的、改變安全性和/或有效性的新信息,F(xiàn)DA可以修改反饋�����。在這種情況下���,F(xiàn)DA將承認(rèn)之前建議發(fā)生了變更,將明確記錄變更的理由�,并將得到適當(dāng)?shù)墓芾硗獾闹С郑c適用的sop相一致���。此外,F(xiàn)DA打算與提交者合作���,解決因變更而提出的任何新問題�,并盡可能考慮到設(shè)備開發(fā)的階段�����。
由于臨床實(shí)踐�����、測(cè)試方法和醫(yī)療器械技術(shù)不斷發(fā)展,F(xiàn)DA建議�����,如果收到FDA的反饋已超過一年�,且研究尚未啟動(dòng),提交者應(yīng)聯(lián)系審查部門���,確認(rèn)FDA之前的建議仍然有效�����。
3、預(yù)審核問題的舉例
3.1有關(guān)監(jiān)管策略問題�����,提交者可以問FDA:
如果我們證明實(shí)質(zhì)等同�����,我們選擇的等同器械是否合適���?
我們希望獲得FDA關(guān)于尋求De Novo請(qǐng)求的反饋和指導(dǎo)�����。我們不知道有任何類似技術(shù)的器械有這種適應(yīng)癥�����,但我們認(rèn)為我們的產(chǎn)品是中等到低風(fēng)險(xiǎn)的�,因此De Novo是合適的。FDA是否知道我們應(yīng)該考慮的其他上市前通知已獲得批準(zhǔn)的器械?FDA是否知道到我們?cè)陲L(fēng)險(xiǎn)評(píng)估中應(yīng)該考慮的其他的技術(shù)問題?
根據(jù)所提供的監(jiān)管策略和臨床前試驗(yàn)討論�,F(xiàn)DA是否同意可能不需要臨床數(shù)據(jù)來支持未來的510(k)試驗(yàn)?
3.2有關(guān)使用適應(yīng)癥/預(yù)期用途問題���,提交者可以問FDA:
FDA對(duì)我們將所述器械稱為OTC有什么擔(dān)心嗎?
使用適應(yīng)癥草案中對(duì)耐藥高血壓的定義是否可接受?
基于預(yù)期用途�����,為新器械提供的尺寸范圍是否合適?
3.3有關(guān)臨床試驗(yàn)問題�����,提交者可以問FDA:
擬議的OUS研究是否足以支持我們的設(shè)備未來的HDE�?
修訂后的臨床研究設(shè)計(jì)�、統(tǒng)計(jì)分析和接受標(biāo)準(zhǔn)是否足以滿足FDA的要求?
主要和次要終點(diǎn)分析是否適用于建議的適應(yīng)證?