第1部分:器械描述和規(guī)范,包括器械的變體版本和附件
預(yù)期用途
技術(shù)文件所包含的器械
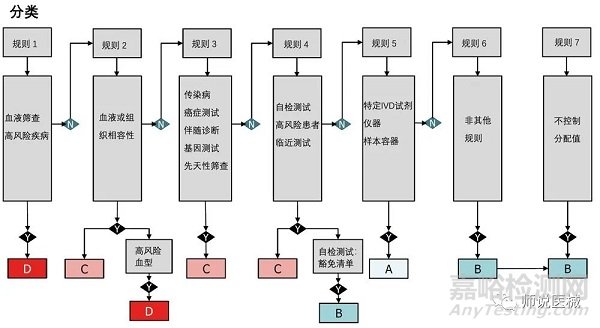
分類
器械描述和規(guī)范說明
器械描述應(yīng)能夠讓人理解器械的設(shè)計(jì)��、構(gòu)成和表現(xiàn)以及其他特征����,同時(shí)應(yīng)包含產(chǎn)品名或商標(biāo)名
應(yīng)提供器械的一般描述��,包括器械的預(yù)期用途和預(yù)期用戶��。
參照前一代和相似代際的器械
第2部分:由制造商提供的信息
標(biāo)簽和使用說明書
第3部分:設(shè)計(jì)與生產(chǎn)信息
材料和組件
系統(tǒng)概述
生產(chǎn)制造信息
參與設(shè)計(jì)和生產(chǎn)活動(dòng)的工廠
第4部分:一般安全性和性能要求(GSPRs)
證明符合GSPRs
產(chǎn)品與設(shè)計(jì)規(guī)范
化學(xué)����、物理和生物學(xué)特性
預(yù)期連接到其他器械才能正常工作的器械
帶測(cè)量功能的器械
輻射防護(hù)
軟件
電氣安全性和電磁兼容性
機(jī)械風(fēng)險(xiǎn)和熱風(fēng)險(xiǎn)防護(hù)
第5部分:受益與風(fēng)險(xiǎn)分析和風(fēng)險(xiǎn)管理
風(fēng)險(xiǎn)管理
第6部分:產(chǎn)品驗(yàn)證與確認(rèn)
樣本類型
性能評(píng)價(jià)與臨床證據(jù)
此處應(yīng)包括可證明以下內(nèi)容的研究:
分析靈敏度
分析特異性
真實(shí)性(偏差)
精確性(可重復(fù)性和可再現(xiàn)性)
準(zhǔn)確性(分析和臨床)
檢測(cè)限和定量
線性度
化驗(yàn)截?cái)?/span>
樣本處理
干擾物質(zhì)(內(nèi)生性和外生性)
交叉反應(yīng)
上市后監(jiān)管和上市后性能跟蹤:
EURL產(chǎn)品驗(yàn)證
第7部分:穩(wěn)定性
穩(wěn)定性包括貨架有效期
包裝和運(yùn)輸驗(yàn)證
滅菌處理
—致性聲明
特定情況中所需的額外信息
伴隨診斷
二�����、IVDR管理體系要求
1.IVD將按照風(fēng)險(xiǎn)高低分為4類

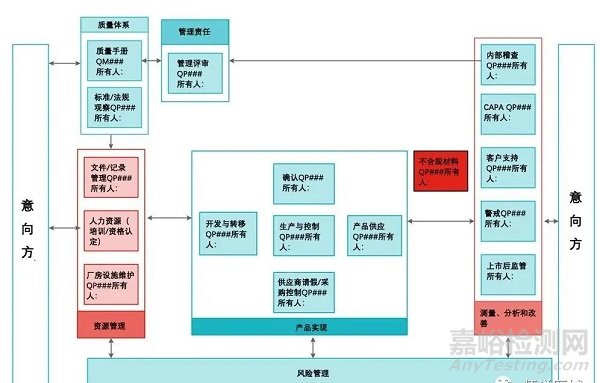
2.管理體系過程圖示例
3.IVDR管理體系
IVDR是客觀看待QMS的一次難得的機(jī)會(huì)����,能確保質(zhì)量管理體系符合商業(yè)目標(biāo)����,同時(shí)實(shí)現(xiàn)合規(guī)性的全部必須要件�����。
具體來說��,在IVDR法規(guī)之下����,QMS必須至少包含:
法規(guī)合規(guī)性策略
適用一般安全性和性能要求的標(biāo)識(shí)
管理責(zé)任
資源管理,包括供應(yīng)商和分包商的選擇與控制
風(fēng)險(xiǎn)管理
性能評(píng)價(jià)����,包括PMPF
產(chǎn)品實(shí)現(xiàn)�����,包括計(jì)劃制定��、設(shè)計(jì)��、開發(fā)�����、生產(chǎn)和服務(wù)提供
驗(yàn)證UDI分配
設(shè)置����、部署并維護(hù)執(zhí)行一整套上市后監(jiān)管系統(tǒng)
負(fù)責(zé)與主管部門��、認(rèn)證機(jī)構(gòu)��、其他經(jīng)濟(jì)經(jīng)營(yíng)實(shí)體�����、客戶和/或其他利益參與方進(jìn)行良好的溝通
警戒管理之下嚴(yán)重事故和現(xiàn)場(chǎng)安全糾正措施的報(bào)告流程
糾正與預(yù)防措施的管理及其有效性驗(yàn)證
輸出����、數(shù)據(jù)分析和產(chǎn)品改善的監(jiān)督和測(cè)量流程
切記��,QMS已落實(shí)到位��,可支持商業(yè)機(jī)構(gòu)實(shí)現(xiàn)其目標(biāo)而不是支持其作為實(shí)體獨(dú)立存在��。
三����、IVDR上市后監(jiān)管要求
根據(jù)第56條和附錄XIII的規(guī)定開展的性能評(píng)價(jià)和上市后監(jiān)管性能評(píng)價(jià)�����,包括新的上市后性能跟蹤(PMPF)計(jì)劃必須在QMS當(dāng)中予以明確定義�����。這是存在差異變化的另外一個(gè)領(lǐng)域�����,需以器械分類為基礎(chǔ)并且應(yīng)當(dāng)予以相應(yīng)的調(diào)整適應(yīng)��。因此����,QMS流程需要有充分的描述信息,同時(shí)允許必要的變化來適應(yīng)制造商不同的器械類型����。PMPF將成為上市后監(jiān)管活動(dòng)的關(guān)鍵要素。計(jì)劃可以是獨(dú)立文件�����,或者在整個(gè)產(chǎn)品系列當(dāng)中都保持充分同質(zhì)性�����,可以在QMS當(dāng)中作為一項(xiàng)操作程序或者一套模板為各產(chǎn)品填寫完成�����。如果預(yù)期作為程序使用����,則程序的范圍還需要充分明確地將其識(shí)別為計(jì)劃。計(jì)劃的引入是較為強(qiáng)烈的指征之一�����,表明IVDR正在從響應(yīng)性的PMS(僅依賴于警戒活動(dòng))轉(zhuǎn)變?yōu)楦又鲃?dòng)的體系��。上市后監(jiān)管活動(dòng)必須根據(jù)第78條以及IVDR中的規(guī)定予以確立、執(zhí)行和維護(hù)��,而且必須作為QMS不可分割的組成部分(見圖3)����。
盡管上市后監(jiān)管活動(dòng)的詳細(xì)信息并非本文的主題內(nèi)容,但仍必須注意該流程和QMS的其他領(lǐng)域之間存在諸多的聯(lián)系��,在法律法規(guī)中已有相關(guān)規(guī)定��,包括風(fēng)險(xiǎn)管理����、性能評(píng)價(jià)、技術(shù)文件更新��、糾正和預(yù)防措施(CAPA)��、不良事件報(bào)告和過程/產(chǎn)品監(jiān)控等等����。對(duì)于C類和D類的IVD��,制造商應(yīng)制作定期安全性更新報(bào)告(PSUR)�����。
制造商最終還需要利用已有的通用規(guī)范(CS)或者“先進(jìn)性”對(duì)比來證實(shí)產(chǎn)品符合安全性和性能的全部要求。
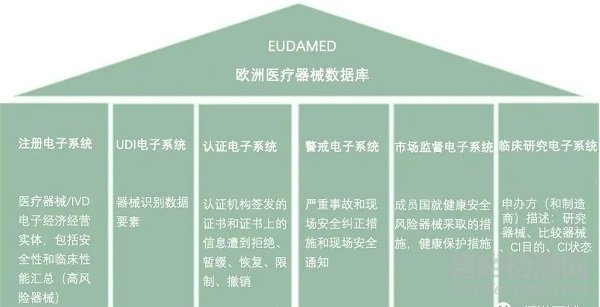
四����、IVDR歐代和注冊(cè)要求
Actors模塊是第一個(gè)需要填寫的模塊。每個(gè)行為人必須輸入明確的數(shù)據(jù)����,如姓名、地址����、網(wǎng)站等。此外�����,識(shí)別和聯(lián)系法規(guī)符合性人員的詳細(xì)信息需要與行為人的數(shù)據(jù)一起輸入����。非歐洲制造商也必須輸入此數(shù)據(jù),因此必須向其所在成員國(guó)的主管當(dāng)局申請(qǐng)SRN�����,并在那里注冊(cè)其LUA。他們的AR需要先在Eudamed注冊(cè)��。對(duì)于初始注冊(cè)�����,無需輸入AR代表的非歐洲制造商的名稱�����。下一步�����,非歐洲制造商在其注冊(cè)過程中指定AR��,之后AR驗(yàn)證該注冊(cè)過程����。
SRN-單一注冊(cè)號(hào)
LUA-本地用戶管理員
五、IVDR合規(guī)建議方案
1.找到合適的分類規(guī)則
2.對(duì)器械進(jìn)行合理的分類

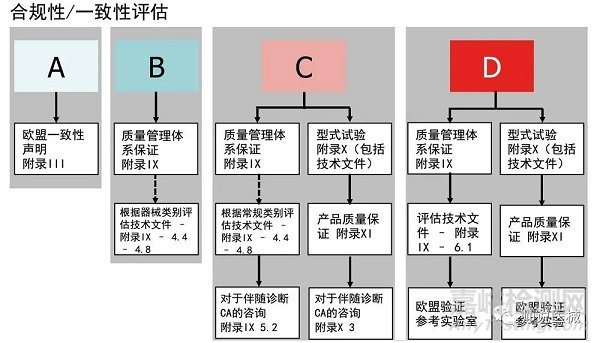
3.合格評(píng)定模式選擇
4.基于自身產(chǎn)品情況尋找合適的顧問公司和轉(zhuǎn)換時(shí)間�����。