距2026年2月2日FDA QMSR體系正式生效僅剩不到2個(gè)月�����,這場(chǎng)替代沿用近30年QSR820法規(guī)的變革�����,已成為全球醫(yī)療器械企業(yè)關(guān)注的核心焦點(diǎn)�。
FDA將于12月16日舉辦的QMSR專(zhuān)題網(wǎng)絡(luò)研討會(huì)�����,倍力醫(yī)療基于研討會(huì)即將介紹的兩大核心板塊(QMSR概覽、QMSR解讀)的PPT與講稿內(nèi)容���,為大家系統(tǒng)梳理核心要點(diǎn),供廣大制造商參考���。
QMSR概述培訓(xùn)-摸清合規(guī)變革的“基本盤(pán)”
QMSR概述培訓(xùn)目標(biāo)的是帶企業(yè)清晰掌握2024年FDA發(fā)布的QMSR最終規(guī)則核心框架�����、QMSR法規(guī)核心定位�����、定義層級(jí)關(guān)系及FDA后續(xù)實(shí)施安排�����,為后續(xù)深度解讀打下基礎(chǔ)�����。
1. 法規(guī)背景:近30年首次重大修訂�����,2026年正式生效
2024年2月2日�,F(xiàn)DA在《聯(lián)邦公報(bào)》發(fā)布該最終規(guī)則,核心是修訂1996年10月制定的21 CFR Part 820�。這是該法規(guī)沿用近30年來(lái)的首次重大修訂�,修訂后法規(guī)名稱(chēng)更新為QMSR�。
值得注意的是���,本次修訂設(shè)置了兩年過(guò)渡期,QMSR正式生效日期為2026年2月2日���,這意味著所有在美國(guó)市場(chǎng)銷(xiāo)售醫(yī)療器械的制造商���,必須在此日期前完成合規(guī)轉(zhuǎn)�����,否則將面臨市場(chǎng)準(zhǔn)入風(fēng)險(xiǎn)�。
2. 核心變革:整合國(guó)際標(biāo)準(zhǔn)���,統(tǒng)一全球合規(guī)框架
QMSR最關(guān)鍵的變革在于通過(guò)“引用納入(IBR)”方式�����,整合了國(guó)際通用標(biāo)準(zhǔn)���,實(shí)現(xiàn)了與全球質(zhì)量管理體系原則的統(tǒng)一。具體來(lái)看:
• 明確援引ISO 13485:2016�����,并非簡(jiǎn)單過(guò)渡或采用該標(biāo)準(zhǔn)�����,而是通過(guò)引用特定條款確保與FDA適用要求保持一致;
• 同時(shí)援引ISO 9000:2015第3條款���,該部分包含應(yīng)用ISO 13485所需的核心術(shù)語(yǔ)和定義�;
• 此舉核心目的是讓全球制造商可采用單一質(zhì)量管理體系�����,高效滿(mǎn)足多國(guó)監(jiān)管要求���,降低跨國(guó)合規(guī)成本�����。
關(guān)于標(biāo)準(zhǔn)獲取�,可通過(guò)ANSI引用標(biāo)準(zhǔn)門(mén)戶(hù)
(ibr.ansi.org/standards/iso1.aspx)查看只讀版本�����,也可通過(guò)該門(mén)戶(hù)購(gòu)買(mǎi)標(biāo)準(zhǔn)副本。
若未來(lái)ISO 13485:2016標(biāo)準(zhǔn)更新���,F(xiàn)DA將評(píng)估其對(duì)本次最終規(guī)則的影響���,必要時(shí)通過(guò)進(jìn)一步規(guī)則制定處理相關(guān)變更�����。
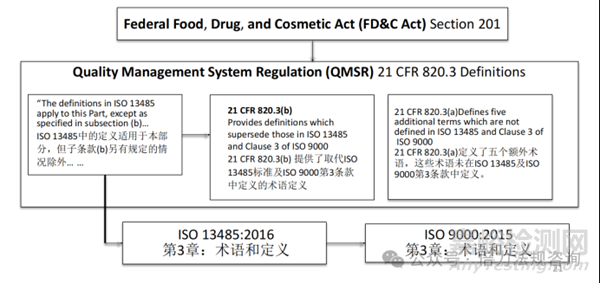
3. 定義層級(jí):清晰界定優(yōu)先級(jí)�,避免合規(guī)混淆
QMSR建立了清晰的定義層級(jí)體系,明確了不同來(lái)源定義的優(yōu)先級(jí)���,這是企業(yè)理解法規(guī)要求的關(guān)鍵:
位于FD&C Act之下�,且在法律層級(jí)中緊隨其后的���,是QMSR 第.3節(jié)所載的法規(guī)及其定義。該法規(guī)同時(shí)闡明了哪些定義優(yōu)先于標(biāo)準(zhǔn)中的定義���,以及哪些術(shù)語(yǔ)雖未在標(biāo)準(zhǔn)中定義�,但由FDA予以界定���。
理解不同定義的排列方式及其層級(jí)關(guān)系至關(guān)重要�����。
在層級(jí)頂端是《食品�、藥品和化妝品法案》(FD&C法案)�����,即法律文本�����。
FD&C 法的第 201 條包含了對(duì)質(zhì)量管理體系要求(QMSR)有影響的定義�����,比如第 201(h)條�,這就是對(duì)醫(yī)療器械的定義���。
在最后,呈現(xiàn)的是標(biāo)準(zhǔn)本身所包含的定義�����。這些定義包括ISO 13485:2016標(biāo)準(zhǔn)第3條款中的術(shù)語(yǔ)與定義,以及ISO 9000:2015標(biāo)準(zhǔn)第3條款中的術(shù)語(yǔ)與定義���。這些定義與標(biāo)準(zhǔn)本身一樣�����,均通過(guò)引用方式納入其中�。
4. FDA實(shí)施安排:保留檢查權(quán)限���,多維度推進(jìn)落地
為確保QMSR順利實(shí)施�,F(xiàn)DA已明確多項(xiàng)核心活動(dòng)�,其中企業(yè)最需關(guān)注的是檢查相關(guān)規(guī)則:
• FDA不會(huì)因檢查結(jié)果向符合ISO 13485:2016標(biāo)準(zhǔn)的企業(yè)頒發(fā)合規(guī)證書(shū)�����;
• 即便持有ISO 13485:2016合規(guī)證書(shū)�,制造商仍需接受FDA常規(guī)檢查���;
• FDA不會(huì)強(qiáng)制要求企業(yè)提供ISO 13485證書(shū)���,核心關(guān)注實(shí)際合規(guī)情況�����。
除此之外���,F(xiàn)DA還將推進(jìn)技術(shù)系統(tǒng)更新、修訂或制定受本規(guī)則影響的合規(guī)計(jì)劃�����、指導(dǎo)文件�����、標(biāo)準(zhǔn)操作規(guī)程等文件�、開(kāi)展內(nèi)部人員培訓(xùn)���、發(fā)布公開(kāi)聲明及外部教育活動(dòng)���,全方位保障法規(guī)落地。
QMSR解讀培訓(xùn)-抓準(zhǔn)合規(guī)實(shí)操的“關(guān)鍵點(diǎn)”
解讀模塊聚焦QMSR的核心要求�����、新舊法規(guī)差異及企業(yè)適配方法���,幫大家從“了解框架”過(guò)渡到“掌握實(shí)操”,明確合規(guī)轉(zhuǎn)型的具體路徑���。
1. 立法目的與適用范圍:延續(xù)核心目標(biāo),明確覆蓋場(chǎng)景
QMSR的核心立法目的與1996版法規(guī)一致���,包括:確保企業(yè)持續(xù)生產(chǎn)符合適用要求和規(guī)格的醫(yī)療器械�����、提供實(shí)現(xiàn)質(zhì)量目標(biāo)的框架�、保障成品器械的安全有效性�、確保符合《聯(lián)邦食品�����、藥品和化妝品法案》要求�。
適用范圍上,QMSR覆蓋所有用于人類(lèi)的成品醫(yī)療器械相關(guān)的全流程活動(dòng)���,具體包括設(shè)計(jì)、制造�、包裝、貼標(biāo)���、儲(chǔ)存�����、安裝���、維護(hù)七大核心環(huán)節(jié)。
值得注意的是���,企業(yè)仍可根據(jù)器械使用方式�����、風(fēng)險(xiǎn)等級(jí)�、復(fù)雜性���、生產(chǎn)工藝風(fēng)險(xiǎn)���、企業(yè)規(guī)模等因素�����,靈活建立適配的質(zhì)量管理體系�,并非采用“一刀切”的要求�����。
同時(shí)���,法規(guī)明確了沖突處理原則:若ISO 13485標(biāo)準(zhǔn)與《聯(lián)邦食品�����、藥品和化妝品法案》或其他FDA法規(guī)沖突�,以FDA相關(guān)要求為準(zhǔn)���;外國(guó)制造商進(jìn)口或擬進(jìn)口器械需滿(mǎn)足QMSR要求�����,法規(guī)也規(guī)定了豁免或變通的判定標(biāo)準(zhǔn)���。
2. 關(guān)鍵條款解讀:聚焦核心要求���,明確合規(guī)邊界
QMSR的核心要求集中在A、B兩部分(C至O部分為保留條款�,未來(lái)可能補(bǔ)充內(nèi)容)�����,重點(diǎn)條款解讀如下:
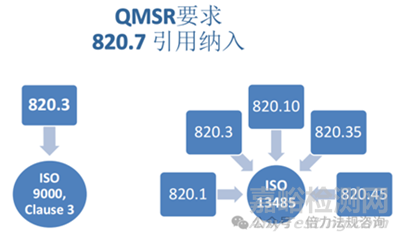
(1)820.7 引用納入(IBR):明確標(biāo)準(zhǔn)適用依據(jù)
該條款正式確認(rèn)通過(guò)引用方式納入ISO 13485:2016及ISO 9000:2015第3條款�����,且所有經(jīng)批準(zhǔn)的引用納入材料可通過(guò)FDA���、美國(guó)國(guó)家檔案和記錄管理局(NARA)及ISO渠道獲取。
其中�����,ISO 13485:2016適用于820.1(范圍)、820.3(定義)���、820.10(質(zhì)量管理體系要求)�����、820.35(記錄控制)�����、820.45(器械標(biāo)簽與包裝控制)等核心章節(jié)。
(2)820.10 質(zhì)量管理體系要求:錨定核心合規(guī)框架
該條款要求企業(yè)建立符合ISO 13485:2016適用要求的文件化質(zhì)量管理體系�����,同時(shí)需滿(mǎn)足其他相關(guān)FDA法規(guī)要求���,包括:
• 21 CFR 830(唯一設(shè)備標(biāo)識(shí)UDI)�;
• 21 CFR 821(醫(yī)療器械追溯要求)�;
• 21 CFR 803(醫(yī)療器械報(bào)告);
• 21 CFR 806(醫(yī)療器械糾正措施與撤回報(bào)告)�。
此外,特別明確II類(lèi)和III類(lèi)器械制造商�,及820.10(c)(1)和(c)(2)表1所列特定I類(lèi)器械制造商�����,需遵守ISO 13485:2016條款7.3的設(shè)計(jì)與開(kāi)發(fā)要求�����;植入器械需滿(mǎn)足該標(biāo)準(zhǔn)條款7.5.9.2關(guān)于維持生命器械的可追溯性要求�。若器械未能滿(mǎn)足第820部分任何適用要求,將被視為“摻假器械”�����,責(zé)任人均會(huì)受到監(jiān)管措施約束。
(3)820.35 記錄控制:明確核心記錄要素
條款要求企業(yè)建立完整的記錄體系�,核心需包含四類(lèi)關(guān)鍵信息:投訴記錄�、維修活動(dòng)記錄、每臺(tái)/每批醫(yī)療器械記錄(含UDI)���、保密信息記錄�����。此舉旨在確保記錄與《聯(lián)邦食品�、藥品和化妝品法案》其他部分保持一致,同時(shí)協(xié)助FDA做好敏感信息的安全處理�����。
(4)820.45 器械標(biāo)簽與包裝控制:強(qiáng)化全流程管控
該條款要求企業(yè)建立并維護(hù)標(biāo)簽與包裝程序的文件記錄�,核心要求包括:在產(chǎn)品放行或儲(chǔ)存前檢查標(biāo)簽與包裝準(zhǔn)確性�����;依據(jù)ISO 13485:2016條款4.2.5記錄標(biāo)簽使用放行文件及檢驗(yàn)結(jié)果;建立操作規(guī)程防止標(biāo)簽與包裝混淆�����,確保標(biāo)簽信息與醫(yī)療器械檔案規(guī)定完全一致�����。
3. 新舊法規(guī)對(duì)比:明確差異點(diǎn)�����,找準(zhǔn)轉(zhuǎn)型重點(diǎn)
1996版QSR820與2024版QMSR核心差異可總結(jié)為以下幾點(diǎn)�����,企業(yè)需重點(diǎn)關(guān)注:
簡(jiǎn)單來(lái)說(shuō)���,QMSR并非顛覆原有框架���,而是在保留核心適用范圍的基礎(chǔ)上���,通過(guò)整合國(guó)際標(biāo)準(zhǔn)實(shí)現(xiàn)合規(guī)框架的統(tǒng)一�,同時(shí)針對(duì)記錄、標(biāo)簽等關(guān)鍵環(huán)節(jié)補(bǔ)充細(xì)化要求�����,強(qiáng)化與其他FDA法規(guī)的銜接。
核心資源匯總:速存這些合規(guī)必備工具
為方便大家后續(xù)深入學(xué)習(xí),整理了本次研討會(huì)提及的核心資源鏈接�����,建議收藏:
• 2024最終規(guī)則原文:
www.federalregister.gov/documents/2024/02/02/2024-01709/medical-devices-quality-system-regulation-amendments
• ISO 13485:2016只讀查看渠道:
ibr.ansi.org/standards/iso1.aspx
• ISO 9000:2015只讀查看渠道:
www.iso.org/obp/ui#iso:std:iso:9000:ed-4:v1:en
• ISO標(biāo)準(zhǔn)副本購(gòu)買(mǎi)渠道:ibr.ansi.org/
組合產(chǎn)品cGMP規(guī)范(21 CFR 4):
www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-4
總結(jié)
本次FDA QMSR專(zhuān)題研討會(huì)的核心邏輯可概括為:發(fā)布《醫(yī)療器械���;質(zhì)量體系法規(guī)修訂》最終規(guī)則(含83條公眾意見(jiàn)及FDA回應(yīng)),通過(guò)IBR方式整合ISO 13485:2016及ISO 9000:2015第3條款�����,建立清晰的定義層級(jí)體系�����;企業(yè)需抓住2026年2月2日這一關(guān)鍵時(shí)間節(jié)點(diǎn)���,通過(guò)FDA提出的七步轉(zhuǎn)型路徑�,實(shí)現(xiàn)從QSR820到QMSR的平穩(wěn)轉(zhuǎn)型。
本次研討會(huì)于中國(guó)時(shí)間凌晨舉行�����,鏈接是:dtps://events.gcc.teams.microsoft.com/event/a5d4dd0d-7f09-4113-a5d9-b49b24ca11f7@7d2fdb41-339c-4257-87f2-a665730b31fc
網(wǎng)絡(luò)研討會(huì)FDA網(wǎng)站:https://www.fda.gov/medical-devices/medical-devices-news-and-events/webinar-quality-management-system-regulation-key-takeaways-12162025